
Signaling beyond Punching Holes: Modulation of Cellular Responses by Vibrio cholerae Cytolysin

Abstract
:1. Introduction
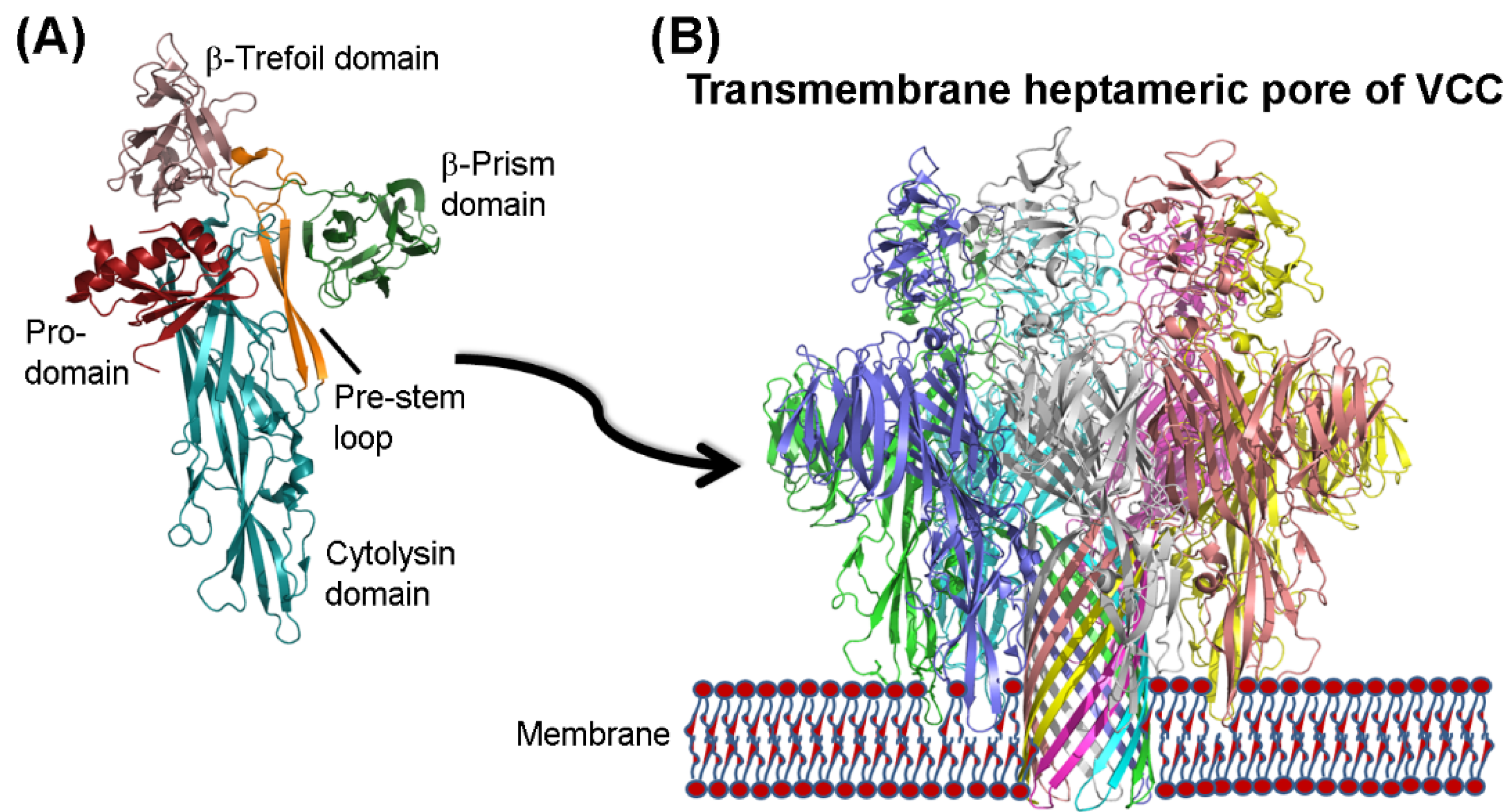
2. Structural Features of VCC


3. Mechanism of Membrane Pore-Formation
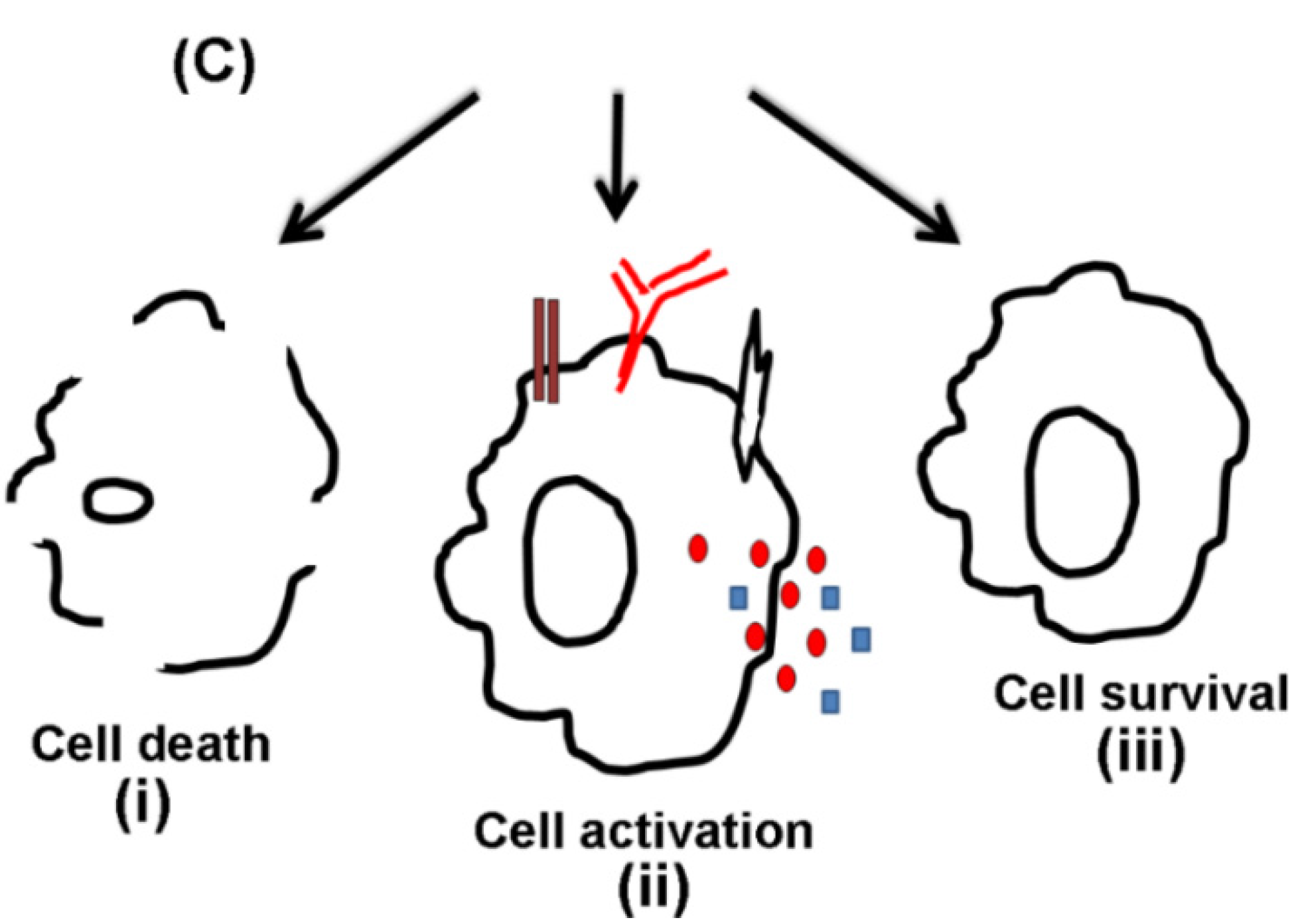
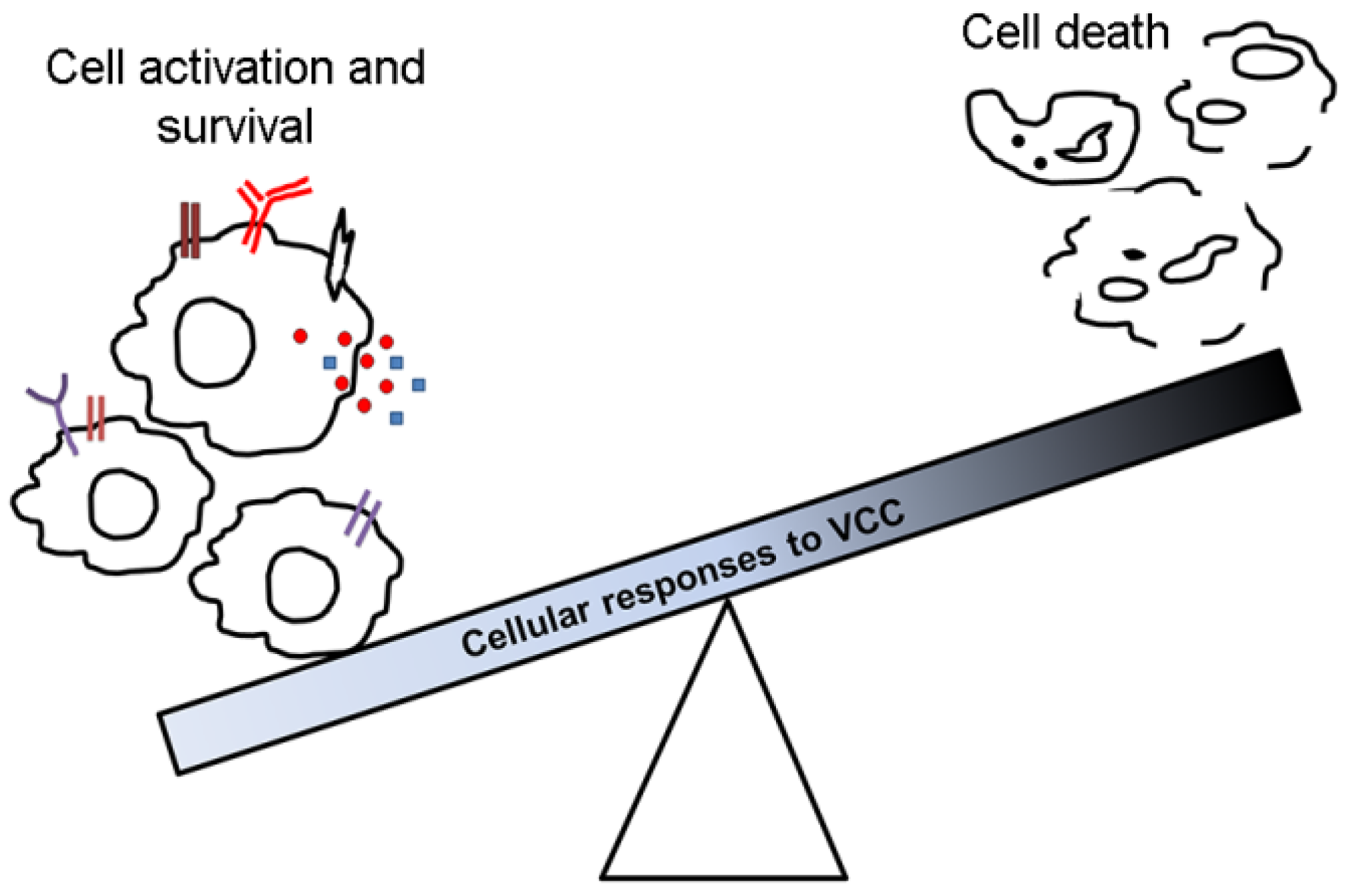
4. Cytotoxic Effects

{kind=link}
{kind=link}
{kind=link}
| Cytotoxic effect | Cell Type | Effects | References |
|---|---|---|---|
| Apoptosis | Int 407 | Efflux of intracellular K+, no intra-nucleosomal degradation, and pores impermeable to Ca2+ | [5] |
| Caco2 and CHO cells | Caspase-3 Activation | [25] | |
| B1a Cells | Caspase-9/-8-dependent apoptosis, lymphocyte apoptosis | [46] | |
| Mouse Peritoneal Macrophages | Caspase-9 dependent apoptosis independent of TLR (Toll-like Receptors) | [47] | |
| Vacuolation | HeLa Cells | VCC from clinical strains of the V. cholerae induces vacuole formation | [48] |
| Vero Cells | VCC present in the culture supernatants of V. cholerae Amazonia causes vacuolation Purified VCC also shows vacuolation effect | [49] | |
| T84 and MDCK-1 | High concentrations of VCC causes damage to the epithelium by stimulating vacuole formation | [50] | |
| Vero cells and BHK cells | Channel formation by VCC is necessary for vacuolating phenotype | [50] |
4.1. Apoptosis Induced by VCC
4.2. Cell Vacuolating Activity
5. Cell Activation by VCC
| Cell Type | Effects | References |
|---|---|---|
| T84 cells | IL-8-dominates inflammatory response, and also upregulation of IL-6 and TNFα | [27] |
| B1a cells | TLR2-dependent NFκB activation and upregulation of CD25 surface expression | [46] |
| Mouse bone marrow derived mast cells | Production of IL-4, IL-5, IL-6 and TNFα and IgE-dependent activation | [62] |
| RAW 264.7 and THP-1 cells | Initiation of pro-inflammatory signaling cascades in response to transmembrane VCC oligomer | [63] |
5.1. Association of VCC with the Pattern Recognition Receptors
5.2. Cytokine Production
6. Cell Survival in Response to VCC
| Cell Type | Effects | References |
|---|---|---|
| Caco2 and CHO | Autophagosome formation and degradation of VCC | [68] |
| HEK | Increased amount of activated LC3 and internalization of VCC by autophagy | [25] |
| C. elegans | Stabilization of HIF-1 inducing hypoxia pathway that leads to enhanced survival | [69] |
| HaCaT cells | Phosphorylation of p38 inducing p38 mediated pathway | [70] |
6.1. Autophagic Response
6.2. p38 Activation
6.3. Hypoxia
7. Conclusions

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaper, J.B.; Morris, J.G., Jr.; Levine, M.M. Cholera. Clin. Microbiol. Rev. 1995, 8, 48–86. [Google Scholar] [PubMed]
- Yamamoto, K.; Ichinose, Y.; Nakasone, N.; Tanabe, M.; Nagahama, M.; Sakurai, J.; Iwanaga, M. Identity of hemolysins produced by Vibrio cholerae non-O1 and V. cholerae O1, biotype El Tor. Infect. Immun. 1986, 51, 927–931. [Google Scholar] [PubMed]
- Saka, H.A.; Bidinost, C.; Sola, C.; Carranza, P.; Collino, C.; Ortiz, S.; Echenique, J.R.; Bocco, J.L. Vibrio cholerae cytolysin is essential for high enterotoxicity and apoptosis induction produced by a cholera toxin gene-negative V. cholerae non-O1, non-O139 strain. Microb. Pathog. 2008, 44, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Al-Omani, M.; Honda, T.; Takeda, Y.; Miwatani, T. Non-O1 Vibrio cholerae hemolysin: Purification, partial characterization, and immunological relatedness to El Tor hemolysin. Infect. Immun. 1984, 45, 192–196. [Google Scholar] [PubMed]
- Zitzer, A.; Wassenaar, T.M.; Walev, I.; Bhakdi, S. Potent membrane-permeabilizing and cytocidal action of Vibrio cholerae cytolysin on human intestinal cells. Infect. Immun. 1997, 65, 1293–1298. [Google Scholar] [PubMed]
- Menzl, K.; Maier, E.; Chakraborty, T.; Benz, R. Hlya hemolysin of V. cholerae O1 biotype El Tor. Eur. J. Biochem. 1996, 240, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, Y.; Yamamoto, K.; Nakasone, N.; Tanabe, M.J.; Takeda, T.; Miwatani, T.; Iwanaga, M. Enterotoxicity of El Tor-like hemolysin of non-O1 V. cholerae. Infect. Immun. 1987, 55, 1090–1093. [Google Scholar] [PubMed]
- Valeva, A.; Walev, I.; Boukhallouk, F.; Wassenaar, T.M.; Heinz, N.; Hedderich, J.; Lautwein, S.; Mocking, M.; Weis, S.; Zitzer, A.; et al. Identification of the membrane penetrating domain of Vibrio cholerae cytolysin as a β-barrel structure. Mol. Microbiol. 2005, 57, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.; Gouaux, E. Vibrio cholerae cytolysin is composed of an alpha-hemolysin-like core. Protein Sci. 2003, 12, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.; Gouaux, E. Crystal structure of the Vibrio cholerae cytolysin (VCC) pro-toxin and its assembly into a heptameric transmembrane pore. J. Mol. Biol. 2005, 350, 997–1016. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Olson, R. Crystal structure of the vibrio cholerae cytolysin heptamer reveals common features among disparate pore-forming toxins. Proc. Natl. Acad. Sci. USA 2011, 108, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, K.; Banerjee, K.K. Unfolding of Vibrio cholerae hemolysin induces oligomerization of the toxin monomer. J. Biol. Chem. 2003, 278, 38470–38475. [Google Scholar] [CrossRef] [PubMed]
- Los, F.C.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef] [PubMed]
- Gouaux, E. Channel-forming toxins: Tales of transformation. Curr. Opin. Struct. Biol. 1997, 7, 566–573. [Google Scholar] [CrossRef]
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Progress Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [CrossRef] [PubMed]
- Bischofberger, M.; Iacovache, I.; van der Goot, F.G. Pathogenic pore-forming proteins: Function and host response. Cell Host Microbe 2012, 12, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Moniatte, M.; van der Goot, F.G.; Buckley, J.T.; Pattus, F.; van Dorsselaer, A. Characterisation of the heptameric pore-forming complex of the aeromonas toxin aerolysin using MALDI-TOF mass spectrometry. FEBS Lett. 1996, 384, 269–272. [Google Scholar] [CrossRef]
- Valeva, A.; Palmer, M.; Bhakdi, S. Staphylococcal alpha-toxin: Formation of the heptameric pore is partially cooperative and proceeds through multiple intermediate stages. Biochemistry 1997, 36, 13298–13304. [Google Scholar] [CrossRef] [PubMed]
- Heuck, A.P.; Tweten, R.K.; Johnson, A.E. Beta-barrel pore-forming toxins: Intriguing dimorphic proteins. Biochemistry 2001, 40, 9065–9073. [Google Scholar] [CrossRef] [PubMed]
- Aroian, R.; Van Der Goot, F. Pore-forming toxins and cellular non-immune defenses (CNIDs). Curr. Opin. Microbiol. 2007, 10, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.K.; Chattopadhyay, K. Vibrio cholerae cytolysin: Structure–function mechanism of an atypical β-barrel pore-forming toxin. In Biochemical Roles of Eukaryotic Cell Surface Macromolecules; Springer International Publishing: Cham, Switzerland, 2015; pp. 109–125. [Google Scholar]
- Krasilnikov, O.V.; Muratkhodjaev, J.N.; Zitzer, A.O. The mode of action of Vibrio cholerae cytolysin. The influences on both erythrocytes and planar lipid bilayers. Biochim. Biophys. Acta (BBA)-Biomembr. 1992, 1111, 7–16. [Google Scholar] [CrossRef]
- Ikigai, H.; Akatsuka, A.; Tsujiyama, H.; Nakae, T.; Shimamura, T. Mechanism of membrane damage by El Tor hemolysin of Vibrio cholerae O1. Infect. Immun. 1996, 64, 2968–2973. [Google Scholar] [PubMed]
- Huntley, J.S.; Sathyamoorthy, V.; Hall, R.H.; Hall, A.C. Membrane attack induced by hlyA, a pore-forming toxin of Vibrio cholerae. Hum. Exp. Toxicol. 1997, 16, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Saka, H.A.; Gutiérrez, M.G.; Bocco, J.L.; Colombo, M.I. The autophagic pathway: A cell survival strategy against the bacterial pore-forming toxin Vibrio cholerae cytolysin. Autophagy 2007, 3, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Bellier, A.; Chen, C.-S.; Kao, C.-Y.; Cinar, H.N.; Aroian, R.V. Hypoxia and the hypoxic response pathway protect against pore-forming toxins in C. elegans. PLoS Pathog. 2009, 5, e1000689. [Google Scholar] [CrossRef] [PubMed]
- Ou, G.; Rompikuntal, P.K.; Bitar, A.; Lindmark, B.; Vaitkevicius, K.; Wai, S.N.; Hammarström, M.-L. Vibrio cholerae cytolysin causes an inflammatory response in human intestinal epithelial cells that is modulated by the prtV protease. PLoS ONE 2009, 4, e7806. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.L.; Murphy, J.R. Molecular cloning of the hemolysin determinant from Vibrio cholerae El Tor. J. Bacteriol. 1984, 160, 239–244. [Google Scholar] [PubMed]
- Yamamoto, K.; Ichinose, Y.; Shinagawa, H.; Makino, K.; Nakata, A.; Iwanaga, M.; Honda, T.; Miwatani, T. Two-step processing for activation of the cytolysin/hemolysin of Vibrio cholerae O1 biotype El Tor: Nucleotide sequence of the structural gene (hlyA) and characterization of the processed products. Infect. Immun. 1990, 58, 4106–4116. [Google Scholar] [PubMed]
- Nagamune, K.; Yamamoto, K.; Honda, T. Intramolecular chaperone activity of the pro-region of Vibrio cholerae El Tor cytolysin. J. Biol. Chem. 1997, 272, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.; Chattopadhyay, K. Unfolding distinguishes the Vibrio cholerae cytolysin precursor from the mature form of the toxin. Biochemistry 2011, 50, 3936–3945. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.; Chattopadhyay, K. Single point mutation in Vibrio cholerae cytolysin compromises the membrane pore-formation mechanism of the toxin. FEBS J. 2012, 279, 4039–4051. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.K.; Chattopadhyay, K. Revisiting the membrane interaction mechanism of a membrane-damaging beta-barrel pore-forming toxin Vibrio cholerae cytolysin. Mol. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.; Chattopadhyay, K. Pre-pore oligomer formation by Vibrio cholerae cytolysin: Insights from a truncated variant lacking the pore-forming pre-stem loop. Biochem. Biophys. Res. Commun. 2014, 443, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.K.; Paul, K.; Chattopadhyay, K. Functional mapping of the lectin activity site on the β-prism domain of Vibrio cholerae cytolysin implications for the membrane pore-formation mechanism of the toxin. J. Biol. Chem. 2013, 288, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.K.; Chattopadhyay, K. Trapping of Vibrio cholerae cytolysin in the membrane-bound monomeric state blocks membrane insertion and functional pore formation by the toxin. J. Biol. Chem. 2014, 289, 16978–16987. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Olson, R. Three-dimensional structure of the detergent-solubilized Vibrio cholerae cytolysin (VCC) heptamer by electron cryomicroscopy. J. Struct. Biol. 2010, 169, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, K.; Bhattacharyya, D.; Banerjee, K.K. Vibrio cholerae hemolysin. Implication of amphiphilicity and lipid-induced conformational change for its pore-forming activity. Eur. J. Biochem. 2002, 269, 4351–4358. [Google Scholar] [CrossRef] [PubMed]
- Zitzer, A.; Harris, J.R.; Kemminer, S.E.; Zitzer, O.; Bhakdi, S.; Muething, J.; Palmer, M. Vibrio cholerae cytolysin: Assembly and membrane insertion of the oligomeric pore are tightly linked and are not detectably restricted by membrane fluidity. Biochim. Biophys. Acta (BBA)-Biomembr. 2000, 1509, 264–274. [Google Scholar] [CrossRef]
- Harris, J.R.; Bhakdi, S.; Meissner, U.; Scheffler, D.; Bittman, R.; Li, G.; Zitzer, A.; Palmer, M. Interaction of the Vibrio cholerae cytolysin (VCC) with cholesterol, some cholesterol esters, and cholesterol derivatives: A tem study. J. Struct. Biol. 2002, 139, 122–135. [Google Scholar] [CrossRef]
- Zitzer, A.; Bittman, R.; Verbicky, C.A.; Erukulla, R.K.; Bhakdi, S.; Weis, S.; Valeva, A.; Palmer, M. Coupling of cholesterol and cone-shaped lipids in bilayers augments membrane permeabilization by the cholesterol-specific toxins streptolysin O and Vibrio cholerae cytolysin. J. Biol. Chem. 2001, 276, 14628–14633. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.L.; Smith, D.J.; Lyras, D.; Chakravorty, A.; Rood, J.I. Programmed cellular necrosis mediated by the pore-forming α-toxin from clostridium septicum. PLoS Pathog. 2009, 5, e1000516. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-Rey, M.; Couzon, F.; Boisset, S.; Brown, E.L.; Bes, M.; Benito, Y.; Barbu, E.M.; Vazquez, V.; Höök, M.; Etienne, J. Staphylococcus aureus panton-valentine leukocidin causes necrotizing pneumonia. Science 2007, 315, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Keyel, P.A.; Roth, R.; Yokoyama, W.M.; Heuser, J.E.; Salter, R.D. Reduction of streptolysin O (SLO) pore-forming activity enhances inflammasome activation. Toxins 2013, 5, 1105–1118. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, G.; Biswas, A.; Banerjee, K.K.; Biswas, T. Vibrio cholerae hemolysin is apoptogenic to peritoneal B-1a cells but its oligomer shepherd the cells for iga response. Mol. Immunol. 2008, 45, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.C.; Mukherjee, G.; Banerjee, P.; Banerjee, K.K.; Biswas, T. Hemolysin induces Toll-like receptor (TLR)-independent apoptosis and multiple TLR-associated parallel activation of macrophages. J. Biol. Chem. 2011, 286, 34542–34551. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; Figueroa, P.; Mukhopadhyay, A.K.; Shimada, T.; Takeda, Y.; Berg, D.E.; Nair, G.B. Cell vacuolation, a manifestation of the El Tor hemolysin of Vibrio cholerae. Infect. Immun. 2000, 68, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.; Andrade, J.R.; Vicente, A.C.P.; Dirita, V.J. Cytotoxic cell vacuolating activity from Vibrio cholerae hemolysin. Infect. Immun. 2000, 68, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Moschioni, M.; Tombola, F.; Bernard, M.D.; Coelho, A.; Zitzer, A.; Zoratti, M.; Montecucco, C. The Vibrio cholerae haemolysin anion channel is required for cell vacuolation and death. Cell. Microbiol. 2002, 4, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Gerschenson, L.; Rotello, R. Apoptosis: A different type of cell death. FASEB J. 1992, 6, 2450–2455. [Google Scholar] [PubMed]
- Carrero, J.A.; Calderon, B.; Unanue, E.R. Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J. Immunol. 2004, 172, 4866–4874. [Google Scholar] [CrossRef] [PubMed]
- Genestier, A.-L.; Michallet, M.-C.; Prévost, G.; Bellot, G.; Chalabreysse, L.; Peyrol, S.; Thivolet, F.; Etienne, J.; Lina, G.; Vallette, F.M. Staphylococcus aureus panton-valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J. Clin. Investig. 2005, 115, 3117. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Sinha, B.; Domschke, W.; Peters, G.; Schulze-Osthoff, K.; Jänicke, R.U. α-toxin is a mediator of staphylococcus aureus–induced cell death and activates caspases via the intrinsic death pathway independently of death receptor signaling. J. Cell Biol. 2001, 155, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Jonas, D.; Walev, I.; Berger, T.; Liebetrau, M.; Palmer, M.; Bhakdi, S. Novel path to apoptosis: Small transmembrane pores created by staphylococcal alpha-toxin in T-lymphocytes evoke internucleosomal DNA degradation. Infect. Immun. 1994, 62, 1304–1312. [Google Scholar] [PubMed]
- Henics, T.; Wheatley, D.N. Cytoplasmic vacuolation, adaptation and cell death: A view on new perspectives and features. Biol. Cell 1999, 91, 485–498. [Google Scholar] [CrossRef]
- Cover, T.L. The vacuolating cytotoxin of Helicobacter pylori. Mol. Microbiol. 1996, 20, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Fivaz, M.; Glauser, P.E.; Parton, R.G.; van der Goot, F.G. A pore-forming toxin interacts with a GPI-anchored protein and causes vacuolation of the endoplasmic reticulum. J. Cell Biol. 1998, 140, 525–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedrigo, G.V.; Campoy, E.M.; Di Venanzio, G.; Colombo, M.I.; Véscovi, E.G. Serratia marcescens is able to survive and proliferate in autophagic-like vacuoles inside non-phagocytic cells. PLoS ONE 2011, 6, e24054. [Google Scholar] [CrossRef] [PubMed]
- Cinar, H.N.; Kothary, M.; Datta, A.R.; Tall, B.D.; Sprando, R.; Bilecen, K.; Yildiz, F.; McCardell, B. Vibrio cholerae hemolysin is required for lethality, developmental delay, and intestinal vacuolation in Caenorhabditis elegans. PLoS ONE 2010, 5, e11558. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A.; Travers, P.; Walport, M.; Capra, J.D. Immunobiology: The Immune System in Health and Disease; Current Biology Publications: New York, NY, USA, 1999. [Google Scholar]
- Arcidiacono, D.; Odom, S.; Frossi, B.; Rivera, J.; Paccani, S.R.; Baldari, C.T.; Pucillo, C.; Montecucco, C.; de Bernard, M. The Vibrio cholerae cytolysin promotes activation of mast cell (T Helper 2) cytokine production. Cell. Microbiol. 2008, 10, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Khilwani, B.; Mukhopadhaya, A.; Chattopadhyay, K. Transmembrane oligomeric form of Vibrio cholerae cytolysin triggers TLR2/TLR6-dependent pro-inflammatory responses in monocytes and macrophages. Biochem. J. 2014, 466, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Ng, V.H.; Maeda, S.; Rest, R.F.; Karin, M. Anthrolysin O and other gram-positive cytolysins are Toll-like receptor 4 agonists. J. Exp. Med. 2004, 200, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Henneke, P.; Visintin, A.; Morse, S.C.; Martin, V.; Watkins, C.; Paton, J.C.; Wessels, M.R.; Golenbock, D.T.; Malley, R. The apoptotic response to pneumolysin is Toll-like receptor 4 dependent and protects against pneumococcal disease. Infect. Immun. 2005, 73, 6479–6487. [Google Scholar] [CrossRef] [PubMed]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; van der Goot, F.G. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 2006, 126, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.; Higa, N.; Koizumi, Y.; Nakasone, N.; Ogura, Y.; McCoy, A.J.; Franchi, L.; Uematsu, S.; Sagara, J.; Taniguchi, S.I. Pathogenic Vibrio activate NLRP3 inflammasome via cytotoxins and TLR/nucleotide-binding oligomerization domain-mediated NF-κB signaling. J. Immunol. 2010, 184, 5287–5297. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Saka, H.A.; Chinen, I.; Zoppino, F.C.; Yoshimori, T.; Bocco, J.L.; Colombo, M.I. Protective role of autophagy against Vibrio cholerae cytolysin, a pore-forming toxin from V. cholerae. Proc. Natl. Acad. Sci. USA 2007, 104, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shao, Z.; Zhai, Z.; Shen, C.; Powell-Coffman, J.A. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS ONE 2009, 4, e6348. [Google Scholar] [CrossRef] [PubMed]
- Kloft, N.; Busch, T.; Neukirch, C.; Weis, S.; Boukhallouk, F.; Bobkiewicz, W.; Cibis, I.; Bhakdi, S.; Husmann, M. Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem. Biophys. Res. Commun. 2009, 385, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, T. Autophagy: A regulated bulk degradation process inside cells. Biochem. Biophys. Res. Commun. 2004, 313, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Kloft, N.; Neukirch, C.; Bobkiewicz, W.; Veerachato, G.; Busch, T.; von Hoven, G.; Boller, K.; Husmann, M. Pro-autophagic signal induction by bacterial pore-forming toxins. Med. Microbiol. Immunol. 2010, 199, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Zitzer, A.; Walev, I.; Palmer, M.; Bhakdi, S. Characterization of Vibrio cholerae El Tor cytolysin as an oligomerizing pore-forming toxin. Med. Microbiol. Immunol. 1995, 184, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Obata, T.; Brown, G.E.; Yaffe, M.B. MAP kinase pathways activated by stress: The p38 MAPK pathway. Crit. Care Med. 2000, 28, N67–N77. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.L.; Abrami, L.; Sasik, R.; Corbeil, J.; van der Goot, F.G.; Aroian, R.V. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. J. Mol. Biol. 2004, 101, 10995–11000. [Google Scholar] [CrossRef] [PubMed]
- Porta, H.; Cancino-Rodezno, A.; Soberón, M.; Bravo, A. Role of MAPK p38 in the cellular responses to pore-forming toxins. Peptides 2011, 32, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Bischof, L.J.; Kao, C.-Y.; Los, F.C.; Gonzalez, M.R.; Shen, Z.; Briggs, S.P.; van der Goot, F.G.; Aroian, R.V. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008, 4, e1000176. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M.; Dersch, K.; Bobkiewicz, W.; Beckmann, E.; Veerachato, G.; Bhakdi, S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus α-toxin or streptolysin O. Biochem. Biophys. Res. Commun. 2006, 344, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.L.; Bischof, L.J.; Griffitts, J.S.; Aroian, R.V. Pore worms: Using Caenorhabditis elegans to study how bacterial toxins interact with their target host. Int. J. Med. Microbiol. 2004, 293, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khilwani, B.; Chattopadhyay, K. Signaling beyond Punching Holes: Modulation of Cellular Responses by Vibrio cholerae Cytolysin. Toxins 2015, 7, 3344-3358. https://doi.org/10.3390/toxins7083344
Khilwani B, Chattopadhyay K. Signaling beyond Punching Holes: Modulation of Cellular Responses by Vibrio cholerae Cytolysin. Toxins. 2015; 7(8):3344-3358. https://doi.org/10.3390/toxins7083344
Chicago/Turabian StyleKhilwani, Barkha, and Kausik Chattopadhyay. 2015. "Signaling beyond Punching Holes: Modulation of Cellular Responses by Vibrio cholerae Cytolysin" Toxins 7, no. 8: 3344-3358. https://doi.org/10.3390/toxins7083344
APA StyleKhilwani, B., & Chattopadhyay, K. (2015). Signaling beyond Punching Holes: Modulation of Cellular Responses by Vibrio cholerae Cytolysin. Toxins, 7(8), 3344-3358. https://doi.org/10.3390/toxins7083344