
The Ins and Outs of Anthrax Toxin

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
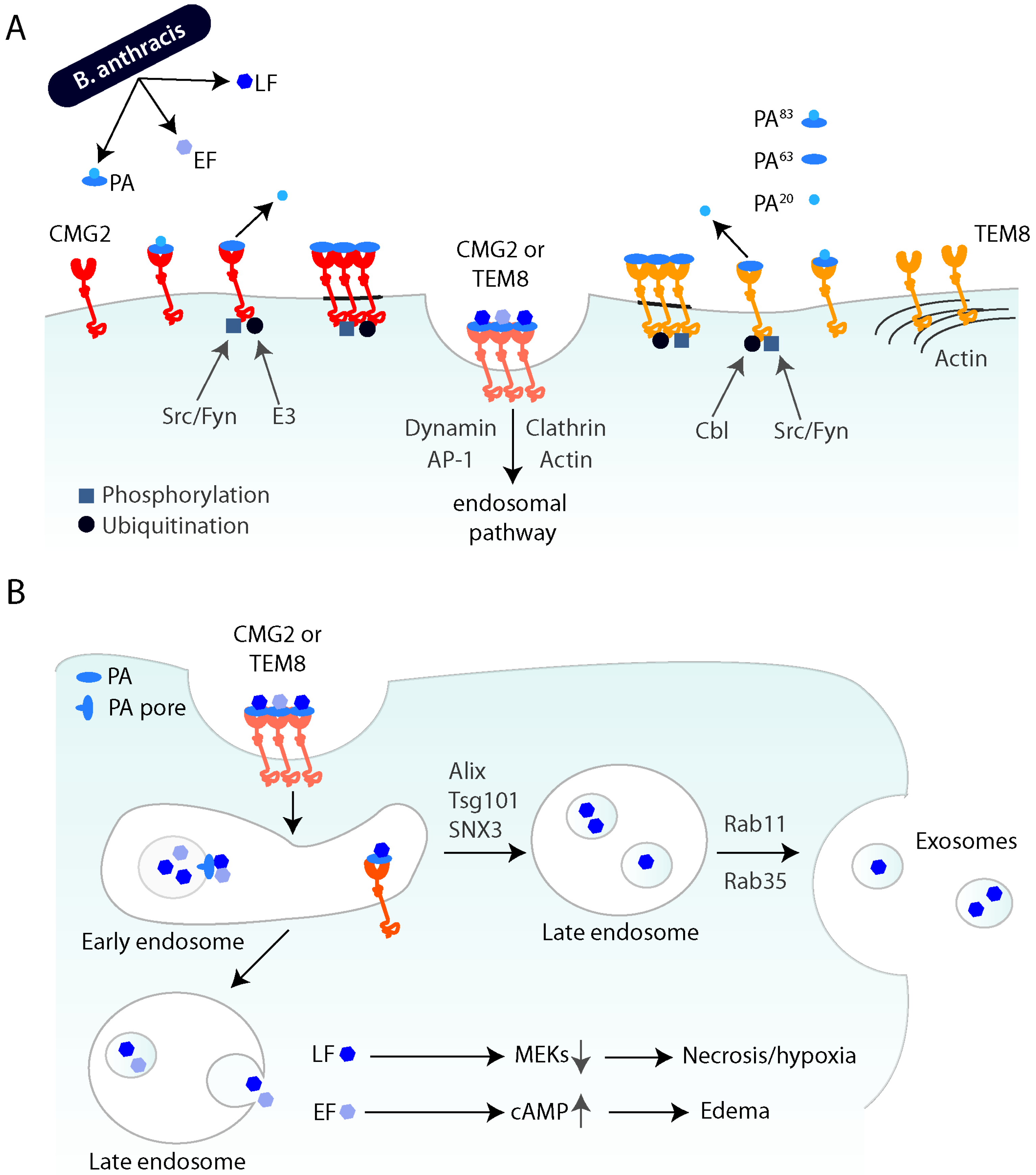
2. Anthrax Toxin Receptors and Toxin Endocytosis
2.1. Cellular Entry and Endocytosis
2.2. Cellular Effects of Anthrax Toxin
3. Toxin Structure
3.1. Protective Antigen
3.2. Binding of PA to Its Cellular Receptor
3.3. PA Pore Structure
3.4. PA Oligomer Diversity
3.5. Lethal Factor Structure
3.6. Edema Factor Structure
3.7. LF/EF Translocation
4. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PA | Protective Antigen |
| LF | Lethal Factor |
| EF | Edema Factor |
| ANTXR1 | Anthrax toxin receptor 1 |
| ANTXR2 | Anthrax toxin receptor 2 |
References
- World Health Organization. International Office of Epizootics, Food, Agriculture Organization of the United Nations. In Anthrax in Humans and Animals, 4th ed.; World Health Organization: Geneva, Switzerland, 2008; p. 208. [Google Scholar]
- Doganay, M.; Metan, G.; Alp, E. A review of cutaneous anthrax and its outcome. J. Infect. Public Health 2010, 3, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, C.N.; Stirling, A.; Smith, J.; Hawkins, G.; Brooks, T.; Hood, J.; Penrice, G.; Browning, L.M.; Ahmed, S.; NHS Greater Glasgow and Clyde; et al. An outbreak of infection with Bacillus anthracis in injecting drug users in Scotland. Eurosurveillance 2010, 15, 19465. [Google Scholar] [PubMed]
- Ringertz, S.H.; Hoiby, E.A.; Jensenius, M.; Maehlen, J.; Caugant, D.A.; Myklebust, A.; Fossum, K. Injectional anthrax in a heroin skin-popper. Lancet 2000, 356, 1574–1575. [Google Scholar] [CrossRef]
- Booth, M.G.; Hood, J.; Brooks, T.J.; Hart, A. Anthrax infection in drug users. Lancet 2010, 375, 1345–1346. [Google Scholar] [CrossRef]
- Hicks, C.W.; Sweeney, D.A.; Cui, X.; Li, Y.; Eichacker, P.Q. An overview of anthrax infection including the recently identified form of disease in injection drug users. Intensive Care Med. 2012, 38, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Bergman, N.H.; Thomason, B.; Shallom, S.; Hazen, A.; Crossno, J.; Rasko, D.A.; Ravel, J.; Read, T.D.; Peterson, S.N.; et al. Formation and composition of the Bacillus anthracis endospore. J. Bacteriol. 2004, 186, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Giorno, R.; Bozue, J.; Cote, C.; Wenzel, T.; Moody, K.S.; Mallozzi, M.; Ryan, M.; Wang, R.; Zielke, R.; Maddock, J.R.; et al. Morphogenesis of the Bacillus anthracis spore. J. Bacteriol. 2007, 189, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Setlow, P. Essential Role of Small, Acid-Soluble Spore Proteins in Resistance of Bacillus-Subtilis Spores to UV-Light. J. Bacteriol. 1986, 167, 174–178. [Google Scholar] [PubMed]
- Hugh-Jones, M.E.; de Vos, V. Anthrax and wildlife. Rev. Sci. Tech. 2002, 21, 359–383. [Google Scholar] [PubMed]
- Goodman, J.W.; Nitecki, D.E. Studies on the relation of a prior immune response to immunogenicity. Immunology 1967, 13, 577–583. [Google Scholar] [PubMed]
- Makino, S.; Watarai, M.; Cheun, H.; Shirahata, T.; Uchida, I. Effect of the lower molecular capsule released from the cell surface of Bacillus anthracis on the pathogenesis of anthrax. J. Infect. Dis. 2002, 186, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Uchida, I.; Terakado, N.; Sasakawa, C.; Yoshikawa, M. Molecular Characterization and Protein-Analysis of the Cap Region, Which Is Essential for Encapsulation in Bacillus-Anthracis. J. Bacteriol. 1989, 171, 722–730. [Google Scholar] [PubMed]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A.T. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Rainey, G.J.A.; Bradley, K.A.; Young, J.A.T. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef] [PubMed]
- Deuquet, J.; Lausch, E.; Superti-Furga, A.; van der Goot, F.G. The dark sides of capillary morphogenesis gene 2. EMBO J. 2012, 31, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.E.; Mavila, A.; Salazar, R.; Bayless, K.J.; Kanagala, S.; Maxwell, S.A.; Davis, G.E. Differential gene expression during capillary morphogenesis in 3D collagen matrices: Regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 2001, 114, 2755–2773. [Google Scholar] [PubMed]
- Hotchkiss, K.A.; Basile, C.M.; Spring, S.C.; Bonuccelli, G.; Lisanti, M.P.; Terman, B.I. TEM8 expression stimulates endothelial cell adhesion and migration by regulating cell-matrix interactions on collagen. Exp. Cell Res. 2005, 305, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.; Kowalczyk, A.P.; Faundez, V. Anthrax toxin receptor 1/tumor endothelium marker 8 mediates cell spreading by coupling extracellular ligands to the actin cytoskeleton. J. Biol. Chem. 2006, 281, 23227–23236. [Google Scholar] [CrossRef] [PubMed]
- Cullen, M.; Seaman, S.; Chaudhary, A.; Yang, M.Y.; Hilton, M.B.; Logsdon, D.; Haines, D.C.; Tessarollo, L.; St Croix, B. Host-derived tumor endothelial marker 8 promotes the growth of melanoma. Cancer Res. 2009, 69, 6021–6026. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.E.; Zhang, Y.; Molinolo, A.A.; Miller-Randolph, S.; Szabo, R.; Bugge, T.H.; Leppla, S.H.; Liu, S. Capillary morphogenesis protein-2 is required for mouse parturition by maintaining uterine collagen homeostasis. Biochem. Biophys. Res. Commun. 2012, 422, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Reeves, C.V.; Wang, X.; Charles-Horvath, P.C.; Vink, J.Y.; Borisenko, V.Y.; Young, J.A.; Kitajewski, J.K. Anthrax toxin receptor 2 functions in ECM homeostasis of the murine reproductive tract and promotes MMP activity. PLoS ONE 2012, 7, e34862. [Google Scholar] [CrossRef] [PubMed]
- Martchenko, M.; Jeong, S.Y.; Cohen, S.N. Heterodimeric integrin complexes containing beta1-integrin promote internalization and lethality of anthrax toxin. Proc. Natl. Acad. Sci. USA 2010, 107, 15583–15588. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Kunz, B.; Deuquet, J.; Bafico, A.; Davidson, G.; van der Goot, F.G. Functional interactions between anthrax toxin receptors and the WNT signalling protein LRP6. Cell Microbiol. 2008, 10, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Lu, Q.; Chaudry, G.J.; Leppla, S.H.; Cohen, S.N. The LDL receptor-related protein LRP6 mediates internalization and lethality of anthrax toxin. Cell 2006, 124, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, Y.; Hoover, B.; Leppla, S.H. The Receptors that Mediate the Direct Lethality of Anthrax Toxin. Toxins 2013, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Crown, D.; Miller-Randolph, S.; Moayeri, M.; Wang, H.L.; Hu, H.J.; Morley, T.; Leppla, S.H. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12424–12429. [Google Scholar] [CrossRef] [PubMed]
- Wigelsworth, D.J.; Krantz, B.A.; Christensen, K.A.; Lacy, D.B.; Juris, S.J.; Collier, R.J. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J. Biol. Chem. 2004, 279, 23349–23356. [Google Scholar] [CrossRef] [PubMed]
- Klimpel, K.R.; Molloy, S.S.; Thomas, G.; Leppla, S.H. Anthrax Toxin Protective Antigen Is Activated by a Cell-Surface Protease with the Sequence Specificity and Catalytic Properties of Furin. Proc. Natl. Acad. Sci. USA 1992, 89, 10277–10281. [Google Scholar] [CrossRef] [PubMed]
- Molloy, S.S.; Bresnahan, P.A.; Leppla, S.H.; Klimpel, K.R.; Thomas, G. Human Furin Is a Calcium-Dependent Serine Endoprotease That Recognizes the Sequence Arg-X-X-Arg and Efficiently Cleaves Anthrax Toxin Protective Antigen. J. Biol. Chem. 1992, 267, 16396–16402. [Google Scholar] [PubMed]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, I.I.; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The Protective Antigen Component of Anthrax Toxin Forms Functional Octameric Complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.C.; Furlong, D.; Hanna, P.C.; Wall, J.S.; Collier, R.J. Anthrax Protective Antigen Forms Oligomers during Intoxication of Mammalian-Cells. J. Biol. Chem. 1994, 269, 20607–20612. [Google Scholar] [PubMed]
- Abrami, L.; Liu, S.; Cosson, P.; Leppla, S.H.; van der Goot, F.G. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 2003, 160, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Leppla, S.H.; van der Goot, F.G. Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J. Cell Biol. 2006, 172, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Kunz, B.; van Der Goot, F.G. Anthrax toxin triggers the activation of src-like kinases to mediate its own uptake. Proc. Natl. Acad. Sci. USA 2010, 107, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.A.; Milano, S.K.; Benovic, J.L. Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 2007, 69, 451–482. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Bischofberger, M.; Kunz, B.; Groux, R.; van der Goot, F.G. Endocytosis of the anthrax toxin is mediated by clathrin, actin and unconventional adaptors. PLoS Pathog. 2010, 6, e1000792. [Google Scholar] [CrossRef] [PubMed]
- Boll, W.; Ehrlich, M.; Collier, R.J.; Kirchhausen, T. Effects of dynamin inactivation on pathways of anthrax toxin uptake. Eur. J. Cell Biol. 2004, 83, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Martchenko, M.; Cohen, S.N. Calpain-dependent cytoskeletal rearrangement exploited for anthrax toxin endocytosis. Proc. Natl. Acad. Sci. USA 2013, 110, E4007–E4015. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wei, W.; Kowalski, P.E.; Chang, A.C.Y.; Cohen, S.N. EST-based genome-wide gene inactivation identifies ARAP3 as a host protein affecting cellular susceptibility to anthrax toxin. Proc. Natl. Acad. Sci. USA 2004, 101, 17246–17251. [Google Scholar] [CrossRef] [PubMed]
- Rainey, G.J.; Wigelsworth, D.J.; Ryan, P.L.; Scobie, H.M.; Collier, R.J.; Young, J.A. Receptor-specific requirements for anthrax toxin delivery into cells. Proc. Natl. Acad. Sci. USA 2005, 102, 13278–13283. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zhang, G.; Li, J.; Chen, P.R. Monitoring endocytic trafficking of anthrax lethal factor by precise and quantitative protein labeling. Angew. Chem. Int. Ed. Engl. 2014, 53, 6449–6453. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Lindsay, M.; Parton, R.G.; Leppla, S.H.; van der Goot, F.G. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J. Cell Biol. 2004, 166, 645–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrami, L.; Brandi, L.; Moayeri, M.; Brown, M.J.; Krantz, B.A.; Leppla, S.H.; van der Goot, F.G. Hijacking multivesicular bodies enables long-term and exosome-mediated long-distance action of anthrax toxin. Cell Rep. 2013, 5, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Banks, D.J.; Barnajian, M.; Maldonado-Arocho, F.J.; Sanchez, A.M.; Bradley, K.A. Anthrax toxin receptor 2 mediates Bacillus anthracis killing of macrophages following spore challenge. Cell Microbiol. 2005, 7, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, M.; Heninger, S.; Hutt, J.; Chen, Y.H.; Lyons, C.R.; Koehler, T.M. Capsule synthesis by Bacillus anthracis is required for dissemination in murine inhalation anthrax. EMBO J. 2005, 24, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lee, Y.S.; Soelaiman, S.; Bergson, P.; Lu, D.; Chen, A.; Beckingham, K.; Grabarek, Z.; Mrksich, M.; Tang, W.J. Physiological calcium concentrations regulate calmodulin binding and catalysis of adenylyl cyclase exotoxins. EMBO J. 2002, 21, 6721–6732. [Google Scholar] [CrossRef] [PubMed]
- Lui, S.; Miller-Randolph, S.; Crown, D.; Moayeri, M.; Sastalla, I.; Okugawa, S.; Leppla, S.H. Anthrax Toxin Targeting of Myeloid Cells through the CMG2 Receptor Is Essential for Establishment of Bacillus anthracis Infections in Mice. Cell Host Microbe 2010, 8, 455–462. [Google Scholar]
- Park, J.M.; Greten, F.R.; Li, Z.W.; Karin, M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 2002, 297, 2048–2051. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.G.; Villasmil, R.; Bernardi, J.; Grene, E.; Cardwell, J.; Wu, A.; Alibek, D.; Bailey, C.; Alibek, K. Lethal toxin of Bacillus anthracis causes apoptosis of macrophages. Biochem. Biophys. Res. Commun. 2002, 293, 349–355. [Google Scholar] [CrossRef]
- Reig, N.; Jiang, A.; Couture, R.; Sutterwala, F.S.; Ogura, Y.; Flavell, R.A.; Mellman, I.; van der Goot, F.G. Maturation modulates caspase-1-independent responses of dendritic cells to Anthrax Lethal Toxin. Cell Microbiol. 2008, 10, 1190–1207. [Google Scholar] [CrossRef] [PubMed]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Crown, D.; Newman, Z.L.; Okugawa, S.; Eckhaus, M.; Cataisson, C.; Liu, S.; Sastalla, I.; Leppla, S.H. Inflammasome Sensor Nlrp1b-Dependent Resistance to Anthrax Is Mediated by Caspase-1, IL-1 Signaling and Neutrophil Recruitment. PLoS Pathog. 2010. [Google Scholar] [CrossRef] [PubMed]
- Terra, J.K.; Cote, C.K.; France, B.; Jenkins, A.L.; Bozue, J.A.; Welkos, S.L.; LeVine, S.M.; Bradley, K.A. Cutting Edge: Resistance to Bacillus anthracis Infection Mediated by a Lethal Toxin Sensitive Allele of Nalp1b/Nlrp1b. J. Immunol. 2010, 184, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Glomski, I.J.; Corre, J.P.; Mock, M.; Goossens, P.L. Noncapsulated toxinogenic Bacillus anthracis presents a specific growth and dissemination pattern in naive and protective antigen-immune mice. Infect. Immun. 2007, 75, 4754–4761. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, R.E.; Hodges, D.R.; Klein, F.; Mahlandt, B.G.; Jones, W.I., Jr.; Haines, B.W.; Rhian, M.A.; Walker, J.S. Role of the lymphatics in the pathogenesis of anthrax. J. Infect. Dis. 1965, 115, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M. The Pathogenesis of Anthrax Following the Administration of Spores by the Respiratory Route. J. Pathol. Bacteriol. 1957, 73, 485–494. [Google Scholar] [CrossRef]
- Brittingham, K.C.; Ruthel, G.; Panchal, R.G.; Fuller, C.L.; Ribot, W.J.; Hoover, T.A.; Young, H.A.; Anderson, A.O.; Bavari, S. Dendritic cells endocytose Bacillus anthracis spores: Implications for anthrax pathogenesis. J. Immunol. 2005, 174, 5545–5552. [Google Scholar] [CrossRef] [PubMed]
- Dixon, T.C.; Fadl, A.A.; Koehler, T.M.; Swanson, J.A.; Hanna, P.C. Early Bacillus anthracis macrophage interactions: Intracellular survival and escape. Cell Microbiol. 2000, 2, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Tournier, J.N.; Quesnel-Hellmann, A.; Cleret, A.; Vidal, D.R. Contribution of toxins to the pathogenesis of inhalational anthrax. Cell Microbiol. 2007, 9, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Greten, F.R.; Wong, A.; Westrick, R.J.; Arthur, J.S.C.; Otsu, K.; Hoffmann, A.; Montminy, M.; Karin, M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis—CREB and NF-kappa B as key regulators. Immunity 2005, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Arocho, F.J.; Fulcher, J.A.; Lee, B.; Bradley, K.A. Anthrax oedema toxin induces anthrax toxin receptor expression in monocyte-derived cells. Mol. Microbiol. 2006, 61, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Larabee, J.L.; Maldonado-Arocho, F.J.; Pacheco, S.; France, B.; DeGiusti, K.; Shakir, S.M.; Bradley, K.A.; Ballard, J.D. Glycogen Synthase Kinase 3 Activation Is Important for Anthrax Edema Toxin-Induced Dendritic Cell Maturation and Anthrax Toxin Receptor 2 Expression in Macrophages. Infect. Immun. 2011, 79, 3302–3308. [Google Scholar] [CrossRef] [PubMed]
- Sastalla, I.; Tang, S.X.; Crown, D.; Liu, S.H.; Eckhaus, M.A.; Hewlett, I.K.; Leppla, S.H.; Moayeri, M. Anthrax Edema Toxin Impairs Clearance in Mice. Infect. Immun. 2012, 80, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Dumetz, F.; Jouvion, G.; Khun, H.; Glomski, I.J.; Corre, J.P.; Rougeaux, C.; Tang, W.J.; Mock, M.; Huerre, M.; Goossens, P.L. Noninvasive Imaging Technologies Reveal Edema Toxin as a Key Virulence Factor in Anthrax. Am. J. Pathol. 2011, 178, 2523–2535. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Wilcox-Adelman, S.; Sano, Y.; Tang, W.J.; Collier, R.J.; Park, J.M. Antiinflammatory cAMP signaling and cell migration genes co-opted by the anthrax bacillus. Proc. Natl. Acad. Sci. USA 2008, 105, 6150–6155. [Google Scholar] [CrossRef] [PubMed]
- Szarowicz, S.E.; During, R.L.; Li, W.; Quinn, C.P.; Tang, W.J.; Southwick, F.S. Bacillus anthracis Edema Toxin Impairs Neutrophil Actin-Based Motility. Infect. Immun. 2009, 77, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Paccani, S.R.; Tonello, F.; Patrussi, L.; Capitani, N.; Simonato, M.; Montecucco, C.; Baldari, C.T. Anthrax toxins inhibit immune cell chemotaxis by perturbing chemokine receptor signalling. Cell Microbiol. 2007, 9, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.; Weiss, S.; Altboum, Z.; Schlomovitz, J.; Glinert, I.; Sittner, A.; Shafferman, A.; Kobiler, D. Differential contribution of Bacillus anthracis toxins to pathogenicity in two animal models. Infect. Immun. 2012, 80, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Welkos, S.L.; Vietri, N.J.; Gibbs, P.H. Non-toxigenic derivatives of the Ames strain of Bacillus anthracis are fully virulent for mice: Role of plasmid pX02 and chromosome in strain-dependent virulence. Microb. Pathog. 1993, 14, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Loving, C.L.; Khurana, T.; Osorio, M.; Lee, G.M.; Kelly, V.K.; Stibitz, S.; Merkel, T.J. Role of anthrax toxins in dissemination, disease progression, and induction of protective adaptive immunity in the mouse aerosol challenge model. Infect. Immun. 2009, 77, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Pezard, C.; Berche, P.; Mock, M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun. 1991, 59, 3472–3477. [Google Scholar] [PubMed]
- Hutt, J.A.; Lovchik, J.A.; Drysdale, M.; Sherwood, R.L.; Brasel, T.; Lipscomb, M.F.; Lyons, C.R. Lethal factor, but not edema factor, is required to cause fatal anthrax in cynomolgus macaques after pulmonary spore challenge. Am. J. Pathol. 2014, 184, 3205–3216. [Google Scholar] [CrossRef] [PubMed]
- Firoved, A.M.; Miller, G.F.; Moayeri, M.; Kakkar, R.; Shen, Y.Q.; Wiggins, J.F.; McNally, E.M.; Tang, W.J.; Leppla, S.H. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am. J. Pathol. 2005, 167, 1309–1320. [Google Scholar] [CrossRef]
- Gnade, B.T.; Moen, S.T.; Chopra, A.K.; Peterson, J.W.; Yeager, L.A. Emergence of Anthrax Edema Toxin as a Master Manipulator of Macrophage and B Cell Functions. Toxins 2010, 2, 1881–1897. [Google Scholar] [CrossRef] [PubMed]
- WHO. Anthrax in Humans and Animals, 4th ed.; WHO: Geneva, Switzerland, 2008. [Google Scholar]
- Guichard, A.; Nizet, V.; Bier, E. New insights into the biological effects of anthrax toxins: Linking cellular to organismal responses. Microbes Infect. 2012, 14, 97–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Zhang, Y.; Moayeri, M.; Liu, J.; Crown, D.; Fattah, R.J.; Wein, A.N.; Yu, Z.X.; Finkel, T.; Leppla, S.H. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 2013, 501, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Pentelute, B.L.; Collier, R.J.; Zhou, Z.H. Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature 2015, 521, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.S.; Earhart, C.A.; Murray, D.L.; Novick, R.P.; Schlievert, P.M.; Ohlendorf, D.H. Structure of toxic shock syndrome toxin 1. Biochemistry 1993, 32, 13761–13766. [Google Scholar] [CrossRef] [PubMed]
- Santelli, E.; Bankston, L.A.; Leppla, S.H.; Liddington, R.C. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature 2004, 430, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Wimalasena, D.S.; Cramer, J.C.; Janowiak, B.E.; Juris, S.J.; Melnyk, R.A.; Anderson, D.E.; Kirk, K.L.; Collier, R.J.; Bann, J.G. Effect of 2-Fluorohistidine Labeling of the Anthrax Protective Antigen on Stability, Pore Formation, and Translocation. Biochemistry 2007, 46, 14928–14936. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of Staphylococcal alpha -Hemolysin, a Heptameric Transmembrane Pore. Science 1996, 274, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Olson, R. Crystal structure of the Vibrio cholerae cytolysin heptamer reveals common features among disparate pore-forming toxins. Proc. Natl. Acad. Sci. USA 2011, 108, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A. A Phenylalanine Clamp Catalyzes Protein Translocation through the Anthrax Toxin Pore. Science 2005, 309, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A.; Finkelstein, A.; Collier, R.J. Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J. Mol. Biol. 2006, 355, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Feld, G.K.; Kintzer, A.F.; Tang, I.I.; Thoren, K.L.; Krantz, B.A. Domain Flexibility Modulates the Heterogeneous Assembly Mechanism of Anthrax Toxin Protective Antigen. J. Mol. Biol. 2012, 415, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Kintzer, A.F.; Sterling, H.J.; Tang, I.I.; Abdul-Gader, A.; Miles, A.J.; Wallace, B.A.; Williams, E.R.; Krantz, B.A. Role of the Protective Antigen Octamer in the Molecular Mechanism of Anthrax Lethal Toxin Stabilization in Plasma. J. Mol. Biol. 2010, 399, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Klimpel, K.R.; Singh, Y.; Leppla, S.H. Fusions of anthrax toxin lethal factor to the ADP-ribosylation domain of Pseudomonas exotoxin A are potent cytotoxins which are translocated to the cytosol of mammalian cells. J. Biol. Chem. 1992, 267, 15542–15548. [Google Scholar] [PubMed]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [PubMed]
- Drum, C.L.; Yan, S.Z.; Sarac, R.; Mabuchi, Y.; Beckingham, K.; Bohm, A.; Grabarek, Z.; Tang, W.J. An extended conformation of calmodulin induces interactions between the structural domains of adenylyl cyclase from Bacillus anthracis to promote catalysis. J. Biol. Chem. 2000, 275, 36334–36340. [Google Scholar] [CrossRef] [PubMed]
- Drum, C.L.; Yan, S.-Z.; Bard, J.; Shen, Y.-Q.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W.J. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 2002, 415, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Udho, E.; Wu, Z.; Collier, R.J.; Finkelstein, A. Protein Translocation through Anthrax Toxin Channels Formed in Planar Lipid Bilayers. Biophys. J. 2004, 87, 3842–3849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Finkelstein, A.; Collier, R.J. Evidence that translocation of anthrax toxin’s lethal factor is initiated by entry of its N terminus into the protective antigen channel. Proc. Natl. Acad. Sci. USA 2004, 101, 16756–16761. [Google Scholar] [CrossRef] [PubMed]
- Feld, G.K.; Thoren, K.L.; Kintzer, A.F.; Sterling, H.J.; Tang, I.I.; Greenberg, S.G.; Williams, E.R.; Krantz, B.A. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat. Struct. Mol. Biol. 2010, 17, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A.; Trivedi, A.D.; Cunningham, K.; Christensen, K.A.; Collier, R.J. Acid-induced unfolding of the amino-terminal domains of the lethal and edema factors of anthrax toxin. J. Mol. Biol. 2004, 344, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; de Barsy, T.; Poole, B.; Trouet, A.; Tulkens, P.; van Hoof, F. Commentary. Lysosomotropic agents. Biochem. Pharmacol. 1974, 23, 2495–2531. [Google Scholar] [CrossRef]
- Ménard, A.; Altendorf, K.; Breves, D.; Mock, M.; Montecucco, C. The vacuolar ATPase proton pump is required for the cytotoxicity of Bacillus anthracis lethal toxin. FEBS Lett. 1996, 386, 161–164. [Google Scholar] [CrossRef]
- Friedlander, A.M. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 1986, 261, 7123–7126. [Google Scholar] [PubMed]
- Baillie, L.; Read, T.D. Bacillus anthracis, a bug with attitude! Curr. Opin. Microbiol. 2001, 4, 78–81. [Google Scholar] [CrossRef]
- Dyer, P.D.R.; Shepherd, T.R.; Gollings, A.S.; Shorter, S.A.; Gorringe-Pattrick, M.A.M.; Tang, C.-K.; Cattoz, B.N.; Baillie, L.; Griffiths, P.C.; Richardson, S.C. Disarmed anthrax toxin delivers antisense oligonucleotides and siRNA with high efficiency and low toxicity. J. Control. Release 2015, 220, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Rabideau, A.E.; Liao, X.L.; Akcay, G.; Pentelute, B.L. Translocation of Non-Canonical Polypeptides into Cells Using Protective Antigen. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Liu, S.; Bankston, L.A.; Liddington, R.C.; Leppla, S.H. Selection of anthrax toxin protective antigen variants that discriminate between the cellular receptors TEM8 and CMG2 and achieve targeting of tumor cells. J. Biol. Chem. 2007, 282, 9834–9845. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.; Hilton, M.B.; Seaman, S.; Haines, D.C.; Stevenson, S.; Lemotte, P.K.; Tschantz, W.R.; Zhang, X.M.; Saha, S.; Fleming, T.; et al. TEM8/ANTXR1 blockade inhibits pathological angiogenesis and potentiates tumoricidal responses against multiple cancer types. Cancer Cell 2012, 21, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Netzel-Arnett, S.; Birkedal-Hansen, H.; Leppla, S.H. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000, 60, 6061–6067. [Google Scholar] [PubMed]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting: MMPs as potential targets in malignancy. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Mekkawy, A.H.; Pourgholami, M.H.; Morris, D.L. Involvement of urokinase-type plasminogen activator system in cancer: An overview. Med. Res. Rev. 2014, 34, 918–956. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Aaronson, H.; Mitola, D.J.; Leppla, S.H.; Bugge, T.H. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. Proc. Natl. Acad. Sci. USA 2003, 100, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.E.; Hoover, B.; Cloud, L.G.; Liu, S.; Molinolo, A.A.; Leppla, S.H.; Bugge, T.H. Comparative toxicity and efficacy of engineered anthrax lethal toxin variants with broad anti-tumor activities. Toxicol. Appl. Pharmacol. 2014, 279, 220–229. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friebe, S.; Van der Goot, F.G.; Bürgi, J. The Ins and Outs of Anthrax Toxin. Toxins 2016, 8, 69. https://doi.org/10.3390/toxins8030069
Friebe S, Van der Goot FG, Bürgi J. The Ins and Outs of Anthrax Toxin. Toxins. 2016; 8(3):69. https://doi.org/10.3390/toxins8030069
Chicago/Turabian StyleFriebe, Sarah, F. Gisou Van der Goot, and Jérôme Bürgi. 2016. "The Ins and Outs of Anthrax Toxin" Toxins 8, no. 3: 69. https://doi.org/10.3390/toxins8030069
APA StyleFriebe, S., Van der Goot, F. G., & Bürgi, J. (2016). The Ins and Outs of Anthrax Toxin. Toxins, 8(3), 69. https://doi.org/10.3390/toxins8030069