
Botulinum Neurotoxin Serotype A Recognizes Its Protein Receptor SV2 by a Different Mechanism than Botulinum Neurotoxin B Synaptotagmin

 , , and
, , and 
Abstract
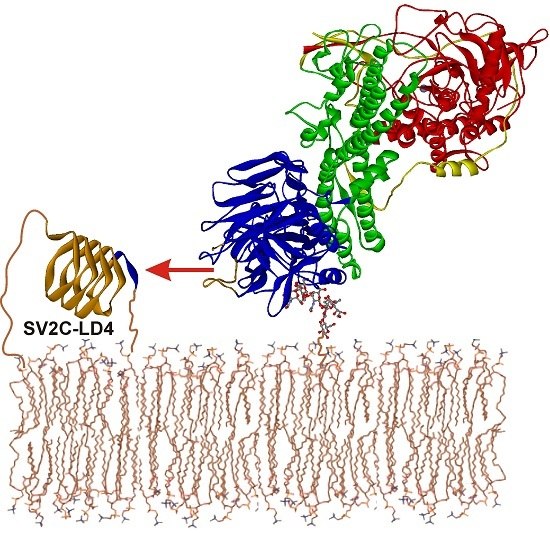
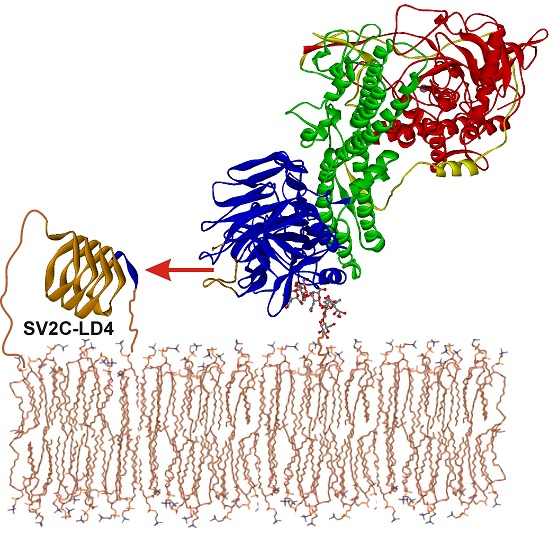
:
1. Introduction
2. Results
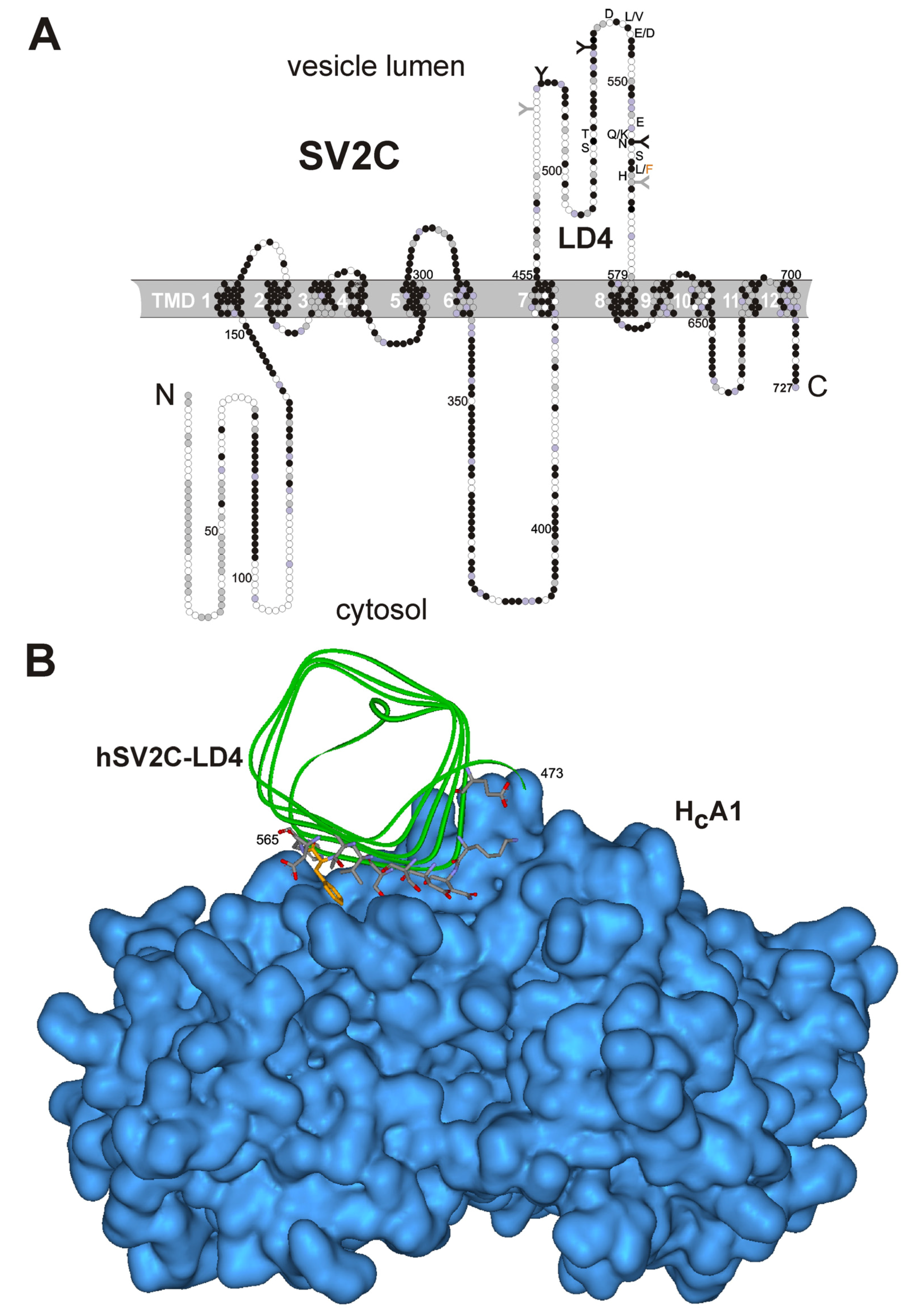
2.1. Effect of Gangliosides and SV2C Transmembrane Domain on Binding of BoNT/A to GST-rSV2C
2.2. Binding of HCA to rSV2A-C Isoforms and hSV2C
2.3. F563L Mutation Decreases Binding of hSV2C to BoNT/A
2.4. Binding Affinity and Kinetics of HCA towards Immobilized GST-hSV2C 454–579
2.5. Intramolecular Blockade of SV2 Binding Site in BoNT/A
2.6. Secondary Structure Analyses of Free and Bound hSV2C Peptide
3. Discussion
3.1. Unlike for BoNT/B–Syt-II, the Dual-Receptor Binding of BoNT/A–SV2C Cannot Be Reconstituted in Solution
3.2. Sequence Diversity in the Interacting Residues and Destabilization of Quadrilateral Helix Might Cause Lower Binding of BoNT/A to SV2A and SV2B as Compared to SV2C
3.3. On a Species Level, Beneficial Mutations Seem to Compensate Loss of Binding Strength Caused by Deteriorating Mutations
3.4. SV2 Peptides Fused to BoNT/A Display a Higher Inhibitory Potency as Syt-II Peptides towards BoNT/B
3.5. The Interaction between HcA and SV2C is Highly Transient, Characterized by Rapid Binding and Unbinding
3.6. A Preformed Structure in Solution and pH-Dependent Binding Distinguish SV2C and Syt-II
4. Conclusions
5. Materials and Methods
5.1. Plasmid Construction
5.2. Expression and Purification of Recombinant Proteins
5.3. Pull Down Assay
5.4. Surface Plasmon Resonance Measurements (SPR)
5.5. Potency of BoNT/A–SV2 Fusion Proteins at MPN Hemidiaphragm Assay
5.6. Circular Dichroism Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AA | amino acid |
| BoNT | botulinum neurotoxin |
| CD | circular dichroism |
| GBS | ganglioside binding site |
| GST | glutathion-S-transferase |
| H | human |
| HC | heavy chain |
| HC | carboxyl-terminal half of HC |
| HCA, HCB, HCE | HC fragment of BoNT serotype A, B and E, respectively |
| HCN and HCC | 25 kDa halves of HC |
| HN | amino-terminal half of HC |
| H6 | N-terminal 6x His tag |
| 6xHN | (HisAsn)6 tag |
| kDa | kilo Dalton |
| L11 | 11-mer linker peptide |
| LC | light chain |
| LD | luminal domain |
| MPN assay | mice phrenic nerve hemidiaphragm assay |
| MW | molecular weight |
| PRP | pentapeptide repeat protein |
| RU | resonance unit |
| S | C-terminal Streptag |
| SDS-PAGE | sodium dodecylsulfate polyacrylamide gel electrophoresis |
| SNAP-25 | synaptosomal associated protein of 25 kDa |
| SNARE | soluble N-ethylmaleimide-sensitive factor attachment protein receptor |
| SPR | surface plasmon resonance |
| SV | synaptic vesicle |
| rSV2A-C | rat synaptic vesicle glycoprotein 2 isoform A, B or C |
| Syt | synaptotagmin |
| TMD | transmembrane domain |
Appendix A


References
- Lamanna, C. Toxicity of bacterial exotoxins by the oral route. Science 1960, 131, 1100–1101. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, H. Botulinum toxin: Application, safety, and limitations. Curr. Top. Microbiol. Immunol. 2013, 364, 307–317. [Google Scholar] [PubMed]
- Lacy, D.B.; Tepp, W.; Cohen, A.C.; DasGupta, B.R.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 1998, 5, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Rummel, A. The long journey of botulinum neurotoxins into the synapse. Toxicon 2015, 107, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Rummel, A. Double receptor anchorage of botulinum neurotoxins accounts for their exquisite neurospecificity. Curr. Top. Microbiol. Immunol. 2013, 364, 61–90. [Google Scholar] [PubMed]
- Fischer, A. Synchronized chaperone function of botulinum neurotoxin domains mediates light chain translocation into neurons. Curr. Top. Microbiol. Immunol. 2013, 364, 115–137. [Google Scholar] [PubMed]
- Blasi, J.; Chapman, E.R.; Link, E.; Binz, T.; Yamasaki, S.; De Camilli, P.; Südhof, T.C.; Niemann, H.; Jahn, R. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 1993, 365, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Santucci, A.; Dasgupta, B.R.; Mehta, P.P.; Jontes, J.; Benfenati, F.; Wilson, M.C.; Montecucco, C. Botulinum neurotoxins serotypes A and E cleave SNAP-25 at distinct COOH-terminal peptide bonds. FEBS Lett. 1993, 335, 99–103. [Google Scholar] [CrossRef]
- Rummel, A.; Mahrhold, S.; Bigalke, H.; Binz, T. The HCC-domain of botulinum neurotoxins A and B exhibits a singular ganglioside binding site displaying serotype specific carbohydrate interaction. Mol. Microbiol. 2004, 51, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Rummel, A.; Häfner, K.; Mahrhold, S.; Darashchonak, N.; Holt, M.; Jahn, R.; Beermann, S.; Karnath, T.; Bigalke, H.; Binz, T. Botulinum neurotoxins C, E and F bind gangliosides via a conserved binding site prior to stimulation-dependent uptake with botulinum neurotoxin F utilising the three isoforms of SV2 as second receptor. J. Neurochem. 2009, 110, 1942–1954. [Google Scholar] [CrossRef] [PubMed]
- Rummel, A.; Eichner, T.; Weil, T.; Karnath, T.; Gutcaits, A.; Mahrhold, S.; Sandhoff, K.; Proia, R.L.; Acharya, K.R.; Bigalke, H.; et al. Identification of the protein receptor binding site of botulinum neurotoxins B and G proves the double-receptor concept. Proc. Natl. Acad. Sci. USA 2007, 104, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, P.; Dupuy, J.; Imamura, A.; Kiso, M.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A in complex with the cell surface co-receptor GT1b-insight into the toxin-neuron interaction. PLoS Pathog. 2008, 4, e1000129. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.A.; Fu, Z.; Kim, J.J.; Baldwin, M.R. Unique ganglioside recognition strategies for clostridial neurotoxins. J. Biol. Chem. 2011, 286, 34015–34022. [Google Scholar] [CrossRef] [PubMed]
- Berntsson, R.P.; Peng, L.; Dong, M.; Stenmark, P. Structure of dual receptor binding to botulinum neurotoxin B. Nat. Commun. 2013, 4, 2058. [Google Scholar] [CrossRef] [PubMed]
- Nishiki, T.; Kamata, Y.; Nemoto, Y.; Omori, A.; Ito, T.; Takahashi, M.; Kozaki, S. Identification of protein receptor for Clostridium botulinum type B neurotoxin in rat brain synaptosomes. J. Biol. Chem. 1994, 269, 10498–10503. [Google Scholar] [PubMed]
- Nishiki, T.; Tokuyama, Y.; Kamata, Y.; Nemoto, Y.; Yoshida, A.; Sekiguchi, M.; Takahashi, M.; Kozaki, S. Binding of botulinum type B neurotoxin to Chinese hamster ovary cells transfected with rat synaptotagmin II cDNA. Neurosci. Lett. 1996, 208, 105–108. [Google Scholar] [CrossRef]
- Nishiki, T.; Tokuyama, Y.; Kamata, Y.; Nemoto, Y.; Yoshida, A.; Sato, K.; Sekiguchi, M.; Takahashi, M.; Kozaki, S. The high-affinity binding of Clostridium botulinum type B neurotoxin to synaptotagmin II associated with gangliosides GT1b/GD1a. FEBS Lett. 1996, 378, 253–257. [Google Scholar] [CrossRef]
- Dong, M.; Richards, D.A.; Goodnough, M.C.; Tepp, W.H.; Johnson, E.A.; Chapman, E.R. Synaptotagmins I and II mediate entry of botulinum neurotoxin B into cells. J. Cell Biol. 2003, 162, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Rummel, A.; Karnath, T.; Henke, T.; Bigalke, H.; Binz, T. Synaptotagmins I and II act as nerve cell receptors for botulinum neurotoxin G. J. Biol. Chem. 2004, 279, 30865–30870. [Google Scholar] [CrossRef] [PubMed]
- Strotmeier, J.; Willjes, G.; Binz, T.; Rummel, A. Human synaptotagmin-II is not a high affinity receptor for botulinum neurotoxin B and G: Increased therapeutic dosage and immunogenicity. FEBS Lett. 2012, 586, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Berntsson, R.P.; Tepp, W.H.; Pitkin, R.M.; Johnson, E.A.; Stenmark, P.; Dong, M. Botulinum neurotoxin D-C uses synaptotagmin I and II as receptors, and human synaptotagmin II is not an effective receptor for type B, D-C and G toxins. J. Cell Sci. 2012, 125, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Yeh, F.; Tepp, W.H.; Dean, C.; Johnson, E.A.; Janz, R.; Chapman, E.R. SV2 is the protein receptor for botulinum neurotoxin A. Science 2006, 312, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Mahrhold, S.; Rummel, A.; Bigalke, H.; Davletov, B.; Binz, T. The synaptic vesicle protein 2C mediates the uptake of botulinum neurotoxin A into phrenic nerves. FEBS Lett. 2006, 580, 2011–2014. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Liu, H.; Tepp, W.H.; Johnson, E.A.; Janz, R.; Chapman, E.R. Glycosylated SV2A and SV2B mediate the entry of botulinum neurotoxin E into neurons. Mol. Biol. Cell 2008, 19, 5226–5237. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Tepp, W.H.; Johnson, E.A.; Dong, M. Botulinum Neurotoxin D Uses Synaptic Vesicle Protein SV2 and Gangliosides as Receptors. PLoS Pathog. 2011, 7, e1002008. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Chen, C.; Barbieri, J.T.; Kim, J.J.; Baldwin, M.R. Glycosylated SV2 and gangliosides as dual receptors for botulinum neurotoxin serotype F. Biochemistry 2009, 48, 5631–5641. [Google Scholar] [CrossRef] [PubMed]
- Ahnert-Hilger, G.; Munster-Wandowski, A.; Holtje, M. Synaptic vesicle proteins: Targets and routes for botulinum neurotoxins. Curr. Top. Microbiol. Immunol. 2013, 364, 159–177. [Google Scholar] [PubMed]
- Janz, R.; Südhof, T.C. SV2C is a synaptic vesicle protein with an unusually restricted localization: Anatomy of a synaptic vesicle protein family. Neuroscience 1999, 94, 1279–1290. [Google Scholar] [CrossRef]
- Benoit, R.M.; Frey, D.; Hilbert, M.; Kevenaar, J.T.; Wieser, M.M.; Stirnimann, C.U.; McMillan, D.; Ceska, T.; Lebon, F.; Jaussi, R.; et al. Structural basis for recognition of synaptic vesicle protein 2C by botulinum neurotoxin A. Nature 2014, 505, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Strotmeier, J.; Mahrhold, S.; Krez, N.; Janzen, C.; Lou, J.; Marks, J.D.; Binz, T.; Rummel, A. Identification of the synaptic vesicle glycoprotein 2 receptor binding site in botulinum neurotoxin A. FEBS Lett. 2014, 588, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Chai, Q.; Arndt, J.W.; Dong, M.; Tepp, W.H.; Johnson, E.A.; Chapman, E.R.; Stevens, R.C. Structural basis of cell surface receptor recognition by botulinum neurotoxin B. Nature 2006, 444, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Rummel, A.; Binz, T.; Brunger, A.T. Botulinum neurotoxin B recognizes its protein receptor with high affinity and specificity. Nature 2006, 444, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Berntsson, R.P.; Peng, L.; Svensson, L.M.; Dong, M.; Stenmark, P. Crystal structures of botulinum neurotoxin DC in complex with its protein receptors synaptotagmin I and II. Structure 2013, 21, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, R.A.; Benoit, R.M. Botulinum neurotoxins: New questions arising from structural biology. Trends Biochem. Sci. 2014, 39, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Mahrhold, S.; Strotmeier, J.; Garcia-Rodriguez, C.; Lou, J.; Marks, J.D.; Rummel, A.; Binz, T. Identification of the SV2 protein receptor-binding site of botulinum neurotoxin type E. Biochem. J. 2013, 453, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Weisemann, J.; Krez, N.; Fiebig, U.; Worbs, S.; Skiba, M.; Endermann, T.; Dorner, M.; Bergström, T.; Munoz, A.; Zegers, I.; et al. Generation and characterization of six recombinant botulinum neurotoxins as reference material to serve in an international proficiency test. Toxins 2015, 7, 5035–5054. [Google Scholar] [CrossRef] [PubMed]
- Weisz, O.A. Acidification and protein traffic. Int. Rev. Cytol. 2003, 226, 259–319. [Google Scholar] [PubMed]
- Bigalke, H.; Rummel, A. Botulinum neurotoxins: Qualitative and quantitative analysis using the mouse phrenic nerve hemidiaphragm assay (MPN). Toxins 2015, 7, 4895–4905. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.S.; Vetting, M.W.; Roderick, S.L.; Mitchenall, L.A.; Maxwell, A.; Takiff, H.E.; Blanchard, J.S. A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 2005, 308, 1480–1483. [Google Scholar] [CrossRef] [PubMed]
- Khrapunov, S.; Cheng, H.; Hegde, S.; Blanchard, J.; Brenowitz, M. Solution structure and refolding of the Mycobacterium tuberculosis pentapeptide repeat protein MfpA. J. Biol. Chem. 2008, 283, 36290–36299. [Google Scholar] [CrossRef] [PubMed]
- Buchko, G.W.; Kim, C.Y.; Terwilliger, T.C.; Kennedy, M.A. Solution structure of the conserved hypothetical protein Rv2302 from Mycobacterium tuberculosis. J. Bacteriol. 2006, 188, 5993–6001. [Google Scholar] [CrossRef] [PubMed]
- Buchko, G.W.; Robinson, H.; Ni, S.; Pakrasi, H.B.; Kennedy, M.A. Cloning, expression, crystallization and preliminary crystallographic analysis of a pentapeptide-repeat protein (Rfr23) from the bacterium Cyanothece 51142. Acta Crystallogr. 2006, 62, 1251–1254. [Google Scholar]
- Buchko, G.W.; Ni, S.; Robinson, H.; Welsh, E.A.; Pakrasi, H.B.; Kennedy, M.A. Characterization of two potentially universal turn motifs that shape the repeated five-residues fold—Crystal structure of a lumenal pentapeptide repeat protein from Cyanothece 51142. Protein Sci. 2006, 15, 2579–2595. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.; Reed, T.A. A set of constructed type spectra for the practical estimation of peptide secondary structure from circular dichroism. Anal. Biochem. 1997, 254, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Lucka, S. Identifikation Essentieller Aminosäuren der Isoform C des Synaptischen Vesikelglykoproteins 2 für die Interaktion Mit Botulinum Neurotoxin Serotyp A; Medizinische Hochschule Hannover: Hannover, Germany, 2011. [Google Scholar]
- Jacky, B.P.; Garay, P.E.; Dupuy, J.; Nelson, J.B.; Cai, B.; Molina, Y.; Wang, J.; Steward, L.E.; Broide, R.S.; Francis, J.; et al. Identification of fibroblast growth factor receptor 3 (FGFR3) as a protein receptor for botulinum neurotoxin serotype A (BoNT/A). PLoS Pathog. 2013, 9, e1003369. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Kita, Y.; Timasheff, S.N. Protein precipitation and denaturation by dimethyl sulfoxide. Biophys. Chem. 2007, 131, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, M.; Wong, K.Y.; Schreiber, G.; Fersht, A.R.; Szabo, A.; Zhou, H.X. Electrostatic enhancement of diffusion-controlled protein-protein association: Comparison of theory and experiment on barnase and barstar. J. Mol. Biol. 1998, 278, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Mushrush, D.J.; Lacy, D.B.; Montal, M. Botulinum neurotoxin devoid of receptor binding domain translocates active protease. PLoS Pathog. 2008, 4, e1000245. [Google Scholar] [CrossRef] [PubMed]
- Binz, T.; Rummel, A. Cell entry strategy of clostridial neurotoxins. J. Neurochem. 2009, 109, 1584–1595. [Google Scholar] [CrossRef] [PubMed]
- Myszka, D.G. Improving biosensor analysis. J. Mol. Recognit. 1999, 12, 279–284. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | pH | ka [M−1s−1] | kd [s−1] | KD Kinetics [M] | KD Steady State [M] |
|---|---|---|---|---|---|
| GST-rSyt-II 1–61 | 7.4 | 5.4 ± 0.3 × 105 | 4.1 ± 0.2 × 10−2 | 7.5 ± 0.8 × 10−8 | 7.2 ± 1.2 × 10−8 |
| GST-rSV2C 454–579 | 7.3 | n.a. | n.a. | n.a. | 5.1 ± 0.3 × 10−7 |
| GST-hSV2C 455–579 | 7.3 | 3.1 ± 0.5 × 105 | 1.2 ± 0.1 × 10−1 | 3.9 ± 0.5 × 10−7 | 4.0 ± 0.6 × 10−7 |
| GST-hSV2C 455–579 | 5.0 | n.a. | n.a. | n.a. | >1.2 × 10−6 |
| GST-hSV2C 455–579 F563L | 7.3 | 1.9 ± 0.1 × 105 | 1.3 ± 0.1 × 10−1 | 6.8 ± 0.1 × 10−7 | 6.6 ± 0.3 × 10−7 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weisemann, J.; Stern, D.; Mahrhold, S.; Dorner, B.G.; Rummel, A. Botulinum Neurotoxin Serotype A Recognizes Its Protein Receptor SV2 by a Different Mechanism than Botulinum Neurotoxin B Synaptotagmin. Toxins 2016, 8, 154. https://doi.org/10.3390/toxins8050154
Weisemann J, Stern D, Mahrhold S, Dorner BG, Rummel A. Botulinum Neurotoxin Serotype A Recognizes Its Protein Receptor SV2 by a Different Mechanism than Botulinum Neurotoxin B Synaptotagmin. Toxins. 2016; 8(5):154. https://doi.org/10.3390/toxins8050154
Chicago/Turabian StyleWeisemann, Jasmin, Daniel Stern, Stefan Mahrhold, Brigitte G. Dorner, and Andreas Rummel. 2016. "Botulinum Neurotoxin Serotype A Recognizes Its Protein Receptor SV2 by a Different Mechanism than Botulinum Neurotoxin B Synaptotagmin" Toxins 8, no. 5: 154. https://doi.org/10.3390/toxins8050154
APA StyleWeisemann, J., Stern, D., Mahrhold, S., Dorner, B. G., & Rummel, A. (2016). Botulinum Neurotoxin Serotype A Recognizes Its Protein Receptor SV2 by a Different Mechanism than Botulinum Neurotoxin B Synaptotagmin. Toxins, 8(5), 154. https://doi.org/10.3390/toxins8050154