
Mapping Protein–Protein Interactions of the Resistance-Related Bacterial Zeta Toxin–Epsilon Antitoxin Complex (ε2ζ2) with High Affinity Peptide Ligands Using Fluorescence Polarization


Abstract
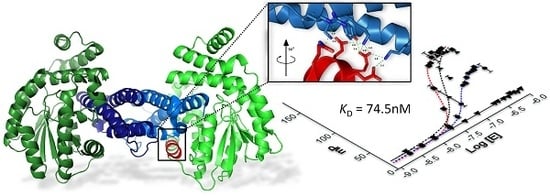
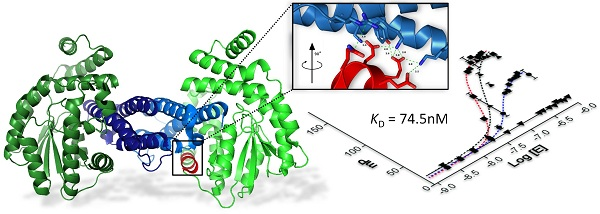
:
1. Introduction
2. Results and Discussion
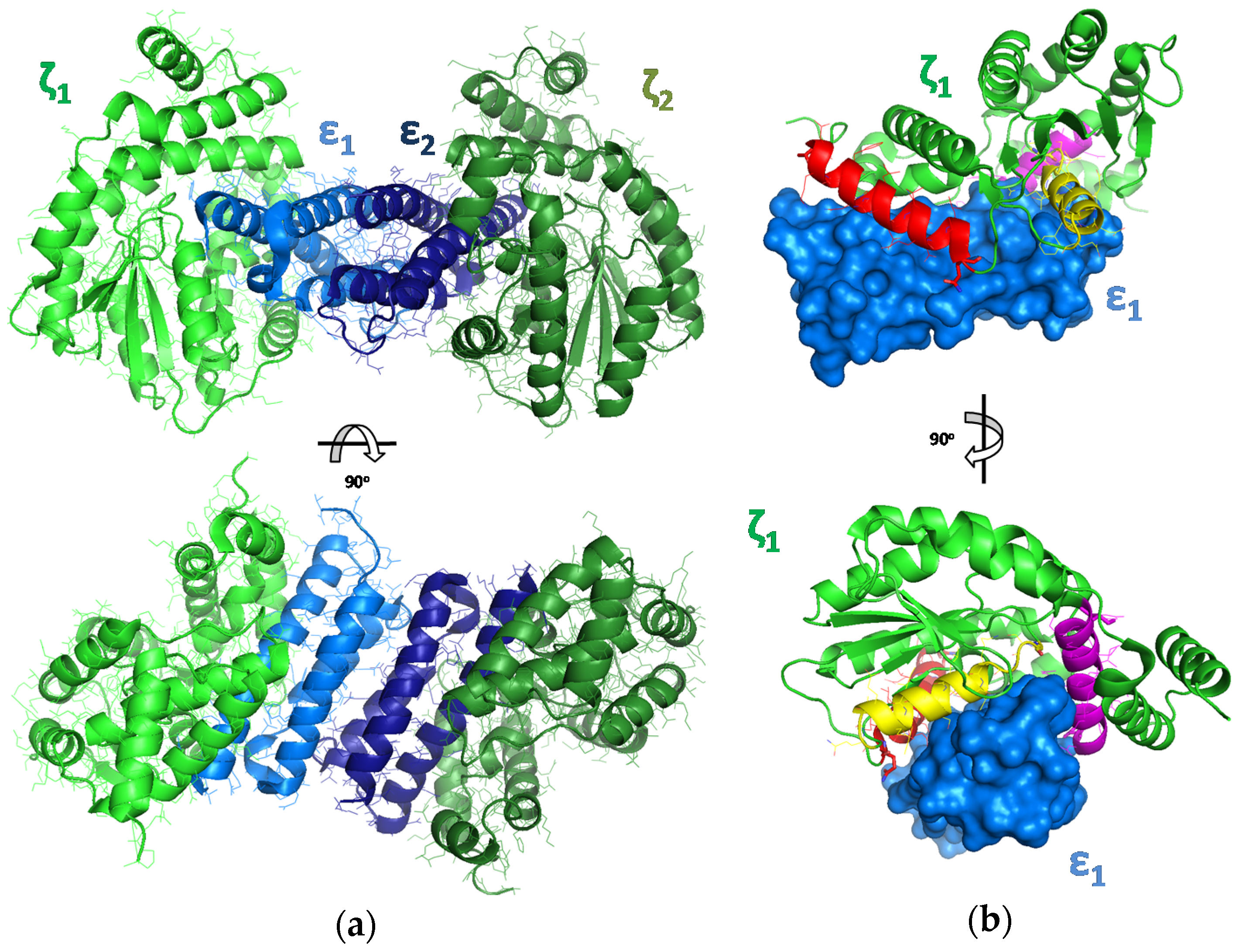
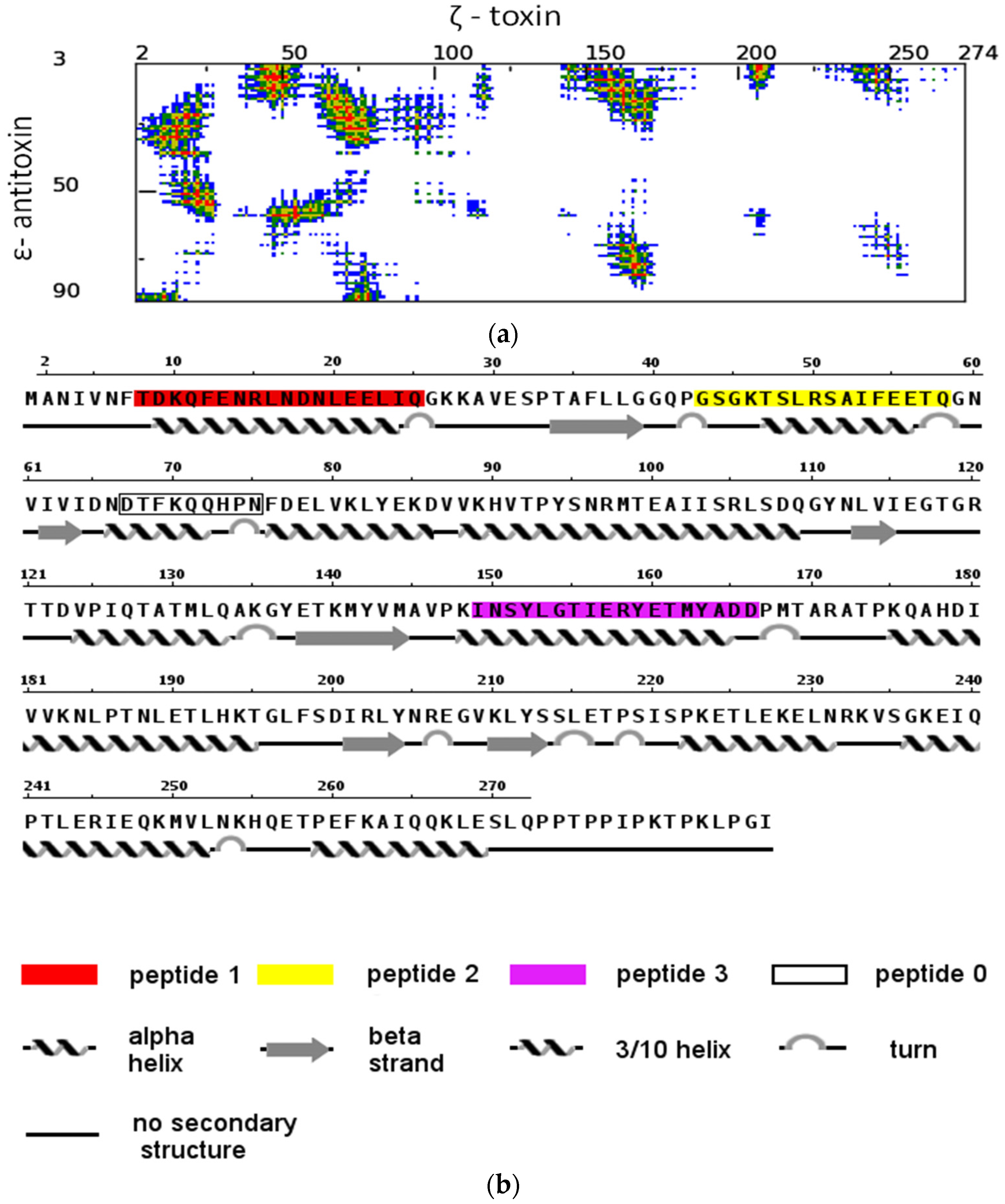
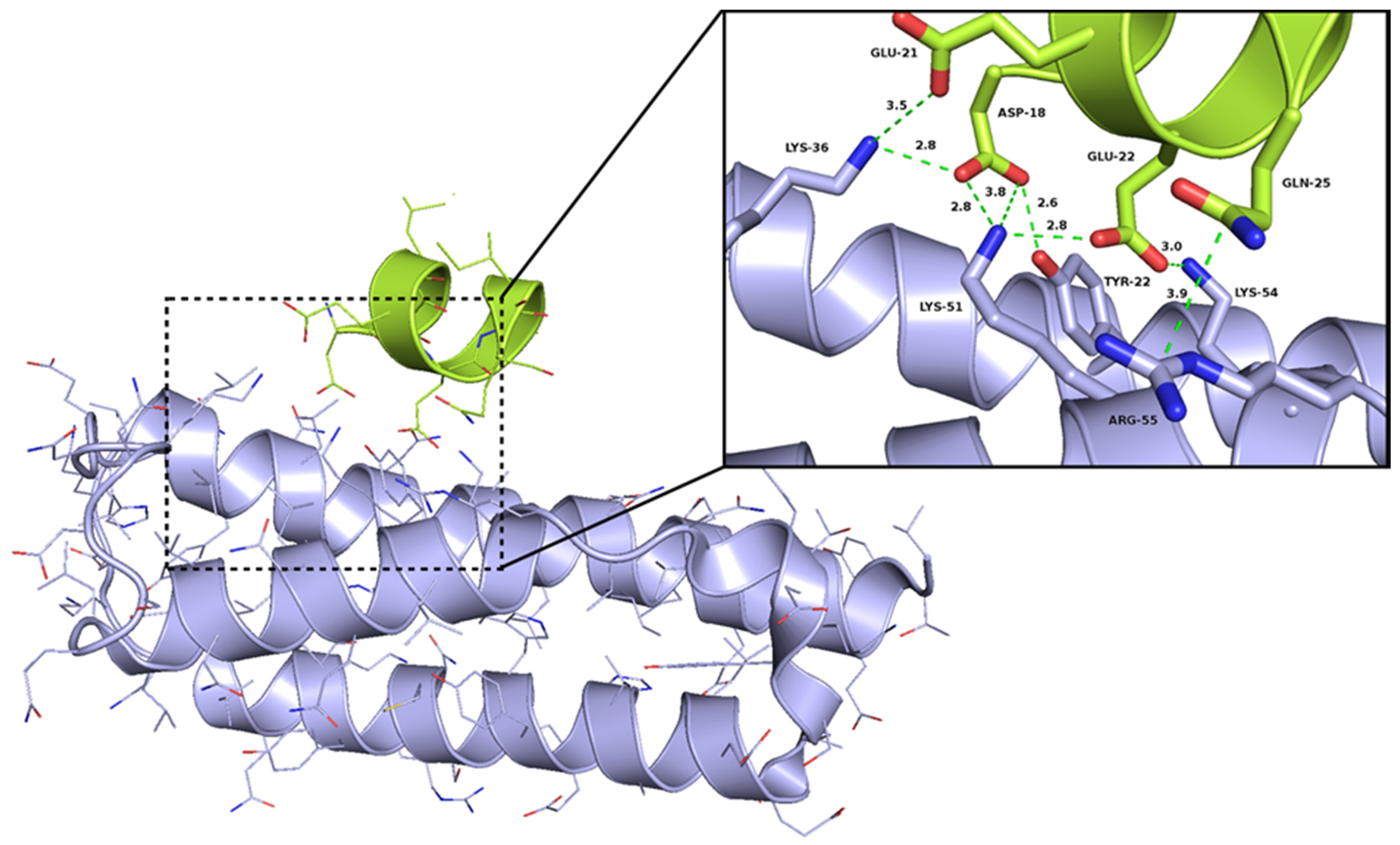
2.1. The Design of Peptides as Potential Disruptors of the Epsilon(ε)–Zeta(ζ) Interaction
2.2. Synthesis of Fluorescein- and Acetyl-Labeled Peptides
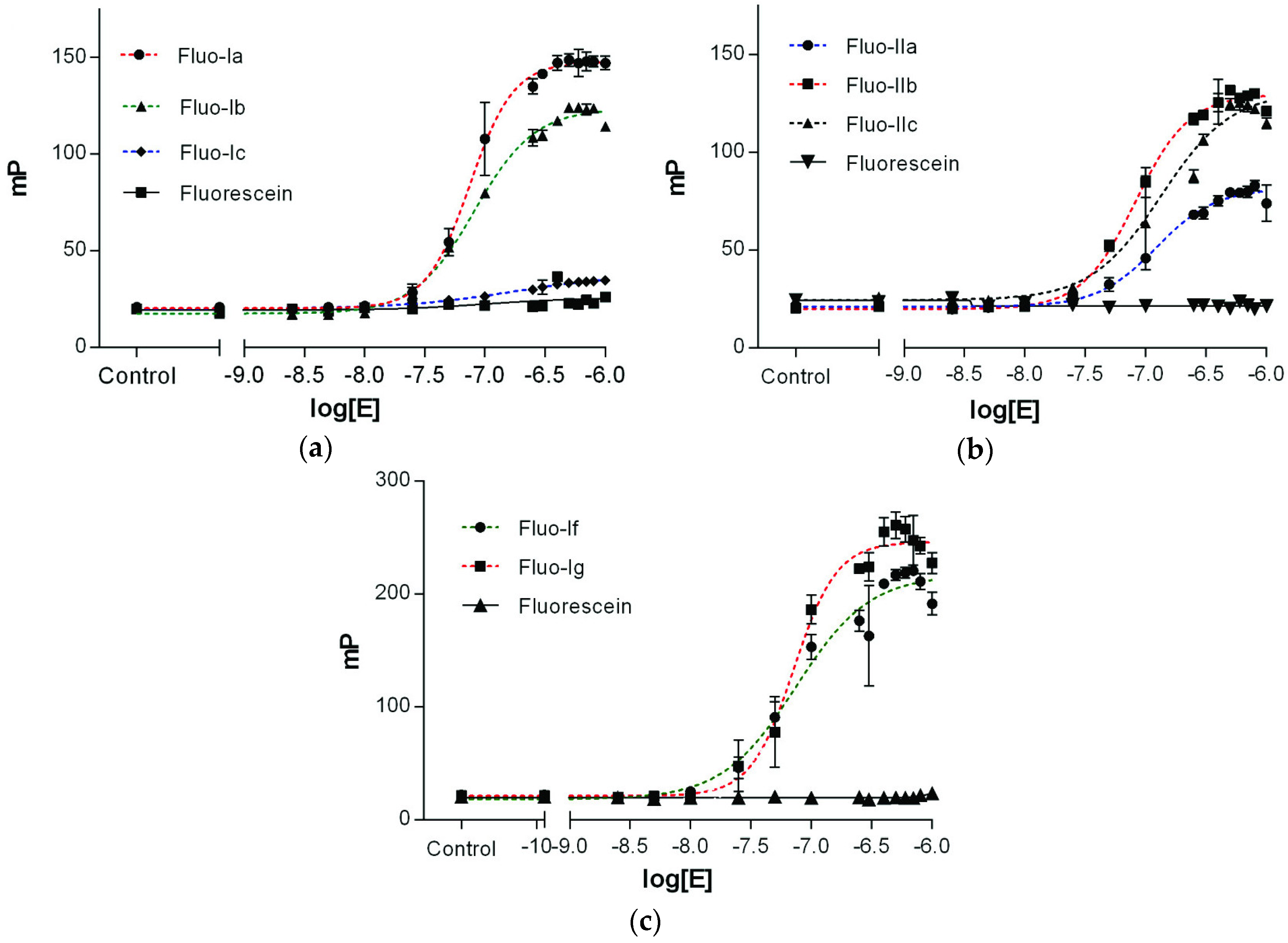
2.3. Characterization of Epsilon–Peptide Interactions by Fluorescence Polarization
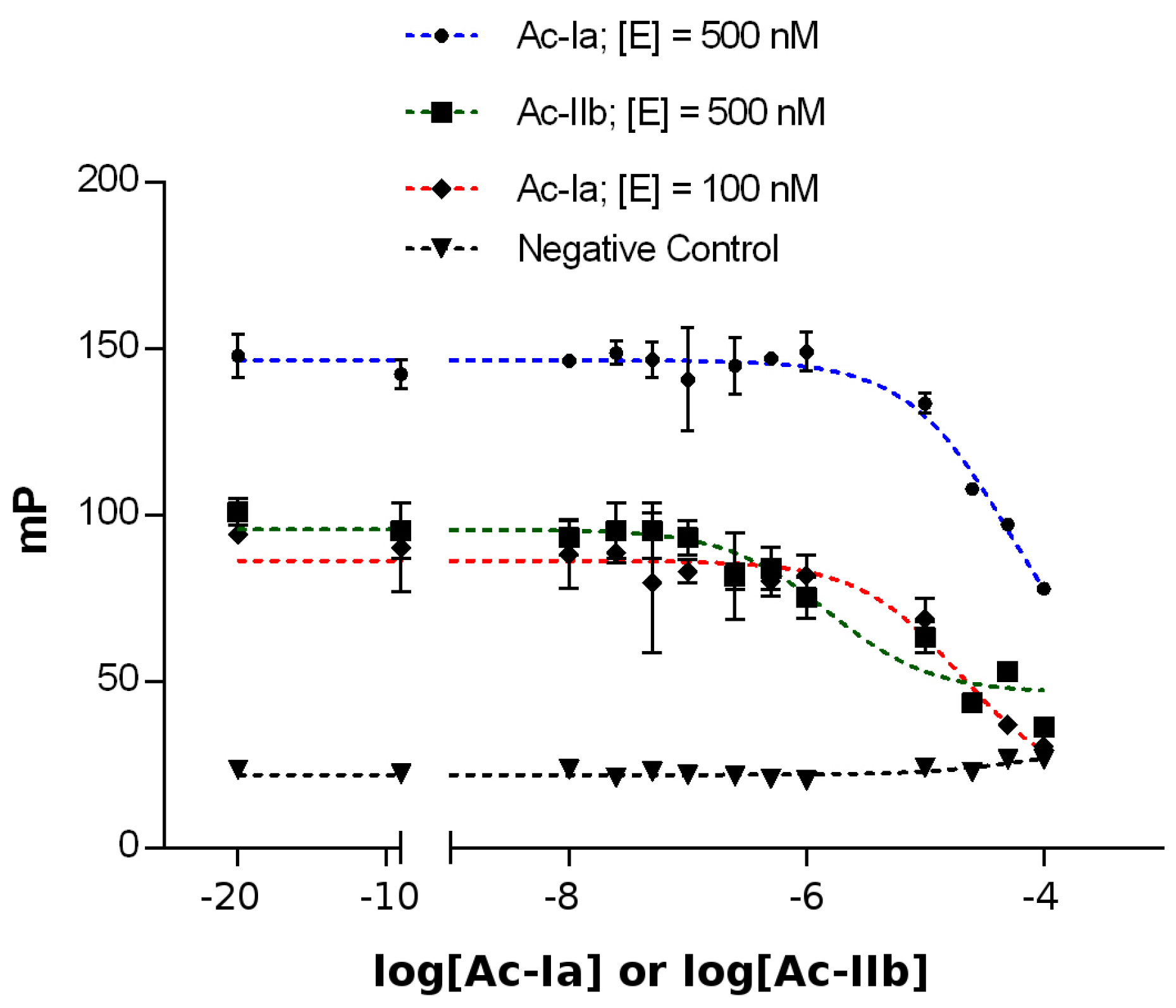
2.4. Competitive Binding Assays
3. Conclusions
4. Materials and Methods
4.1. Peptide Design
4.2. Reagents and Resins
4.3. Synthesis
4.4. Protein Purification
4.5. Fluorescence Polarization Assay
4.6. Competitive Binding Assays
4.7. Calculation of the Z’-Factor
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Michael, C.A.; Dominey-Howes, D.; Labbate, M. The antimicrobial resistance crisis: Causes, consequences, and management. Front. Public Health 2014, 2, 145. [Google Scholar] [CrossRef] [PubMed]
- Roghmann, M.C.; McGrail, L. Novel ways of preventing antibiotic-resistant infections: What might the future hold? Am. J. Infect. Control 2006, 34, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Barriere, S.L. Clinical, economic and societal impact of antibiotic resistance. Expert Opin. Pharmacother. 2015, 16, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Collignon, P.; Athukorala, P.C.; Senanayake, S.; Khan, F. Antimicrobial resistance: The major contribution of poor governance and corruption to this growing problem. PLoS ONE 2015, 10, e0116746. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L.; Baquero, F. Emergence and spread of antibiotic resistance: Setting a parameter space. Upsala J. Med. Sci. 2014, 119, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Collignon, P. Antibiotic resistance: Are we all doomed? Intern. Med. J. 2015, 45, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, V.D. Health-care-associated infections in developing countries. Lancet 2011, 377, 186–188. [Google Scholar] [CrossRef]
- Spellberg, B.; Bartlett, J.; Wunderink, R.; Gilbert, D.N. Novel approaches are needed to develop tomorrow’s antibacterial therapies. Am. J. Respir. Crit. Care Med. 2015, 191, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Powers, J.H.; Brass, E.P.; Miller, L.G.; Edwards, J.E., Jr. Trends in antimicrobial drug development: Implications for the future. Clin. Infect. Dis. 2004, 38, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J. Microbiology. Desperately seeking new antibiotics. Science 2008, 321, 1644–1645. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Donadio, S.; Maffioli, S.; Monciardini, P.; Sosio, M.; Jabes, D. Antibiotic discovery in the twenty-first century: Current trends and future perspectives. J. Antibiot. 2010, 63, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Harrison, E.M.; Bi, D.; Tai, C.; He, X.; Ou, H.Y.; Rajakumar, K.; Deng, Z. TADB: A web-based resource for type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011, 39, D606–D611. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Park, J.H.; Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K. Prokaryotic Toxin-Antitoxins, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Goeders, N.; Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins 2014, 6, 304–324. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.F.; Bertram, R. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 2013, 340, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, S.J.; Poppenberger, B.; Rozhon, W. Toxin-antitoxin systems: Biology, identification, and application. Mob. Genet. Elem. 2013, 3, e26219. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.J.; Hergenrother, P.J. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012, 20, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Nieto, C.; Pellicer, T.; Balsa, D.; Christensen, S.K.; Gerdes, K.; Espinosa, M. The chromosomal relBE2 toxin-antitoxin locus of Streptococcus pneumoniae: Characterization and use of a bioluminescence resonance energy transfer assay to detect toxin-antitoxin interaction. Mol. Microbiol. 2006, 59, 1280–1296. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.T.; Balsa, D.; Espinosa, M. One cannot rule them all: Are bacterial toxins-antitoxins druggable? FEMS Microbiol. Rev. 2015, 39, 522–540. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Mishra, N.K.; Bhatnagar, S.; Bhatnagar, R. PemK toxin of Bacillus anthracis is a ribonuclease: An insight into its active site, structure, and function. J. Biol. Chem. 2010, 285, 7254–7270. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.G.; Lee, S.J.; Chae, S.; Lee, K.Y.; Kim, J.H.; Lee, B.J. Structural and functional studies of the Mycobacterium tuberculosis VapBC30 toxin-antitoxin system: Implications for the design of novel antimicrobial peptides. Nucleic Acids Res. 2015, 43, 7624–7637. [Google Scholar] [CrossRef] [PubMed]
- Lioy, V.S.; Rey, O.; Balsa, D.; Pellicer, T.; Alonso, J.C. A toxin-antitoxin module as a target for antimicrobial development. Plasmid 2010, 63, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Behnke, D.; Malke, H.; Hartmann, M.; Walter, F. Post-transformational rearrangement of an in vitro reconstructed group-A streptococcal erythromycin resistance plasmid. Plasmid 1979, 2, 605–616. [Google Scholar] [CrossRef]
- Camacho, A.G.; Misselwitz, R.; Behlke, J.; Ayora, S.; Welfle, K.; Meinhart, A.; Lara, B.; Saenger, W.; Welfle, H.; Alonso, J.C. In vitro and in vivo stability of the epsilon2zeta2 protein complex of the broad host-range Streptococcus pyogenes pSM19035 addiction system. Biol. Chem. 2002, 383, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Zielenkiewicz, U.; Ceglowski, P. The toxin-antitoxin system of the streptococcal plasmid pSM19035. J. Bacteriol. 2005, 187, 6094–6105. [Google Scholar] [CrossRef] [PubMed]
- Brantl, S.; Behnke, D.; Alonso, J.C. Molecular analysis of the replication region of the conjugative Streptococcus agalactiae plasmid pIP501 in Bacillus subtilis. Comparison with plasmids pAMbeta1 and pSM19035. Nucleic Acids Res. 1990, 18, 4783–4790. [Google Scholar] [CrossRef] [PubMed]
- Holden, M.T.; Hauser, H.; Sanders, M.; Ngo, T.H.; Cherevach, I.; Cronin, A.; Goodhead, I.; Mungall, K.; Quail, M.A.; Price, C.; et al. Rapid evolution of virulence and drug resistance in the emerging zoonotic pathogen Streptococcus suis. PLoS ONE 2009, 4, e6072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, F.V.; Perreten, V.; Teuber, M. Sequence of the 50-kb conjugative multiresistance plasmid pRE25 from Enterococcus faecalis RE25. Plasmid 2001, 46, 170–187. [Google Scholar] [CrossRef] [PubMed]
- Sletvold, H.; Johnsen, P.J.; Hamre, I.; Simonsen, G.S.; Sundsfjord, A.; Nielsen, K.M. Complete sequence of Enterococcus faecium pVEF3 and the detection of an omega-epsilon-zeta toxin-antitoxin module and an ABC transporter. Plasmid 2008, 60, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, H.; Meinhart, A. Epsilon/zeta systems: Their role in resistance, virulence, and their potential for antibiotic development. J. Mol. Med. 2011, 89, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Pachulec, E.; van der Does, C. Conjugative plasmids of Neisseria gonorrhoeae. PLoS ONE 2010, 5, e9962. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; Saavedra De Bast, M. Bacterial toxin-antitoxin systems: More than selfish entities? PLoS Genet. 2009, 5, e1000437. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, H.; Gebhardt, M.; Shoeman, R.L.; Meinhart, A. A novel mechanism of programmed cell death in bacteria by toxin-antitoxin systems corrupts peptidoglycan synthesis. PLoS Biol. 2011, 9, e1001033. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.B.; Gerdes, K. Programmed cell death in bacteria: Proteic plasmid stabilization systems. Mol. Microbiol. 1995, 17, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, I.; Zielenkiewicz, U. The ClpXP protease is responsible for the degradation of the Epsilon antidote to the Zeta toxin of the streptococcal pSM19035 plasmid. J. Biol. Chem. 2014, 289, 7514–7523. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef] [PubMed]
- Saupe, J.; Roske, Y.; Schillinger, C.; Kamdem, N.; Radetzki, S.; Diehl, A.; Oschkinat, H.; Krause, G.; Heinemann, U.; Rademann, J. Discovery, structure-activity relationship studies, and crystal structure of nonpeptide inhibitors bound to the Shank3 PDZ domain. ChemMedChem 2011, 6, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Fuller, J.C.; Burgoyne, N.J.; Jackson, R.M. Predicting druggable binding sites at the protein-protein interface. Drug Discov. Today 2009, 14, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.G.; Guo, J.; Wu, Y. Chemokine receptor CCR5 antagonist maraviroc: Medicinal chemistry and clinical applications. Curr. Top. Med. Chem. 2014, 14, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Domling, A. Small molecular weight protein-protein interaction antagonists: An insurmountable challenge? Curr. Opin. Chem. Biol. 2008, 12, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Li, X.J. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Matthews, J.M. Protein-protein interactions in human disease. Curr. Opin. Struct. Biol. 2005, 15, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angew. Chem. Int. Ed. Engl. 2015, 54, 8896–8927. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Lee, G.I.; Park, H.S.; Payne, G.A.; Rodriguez, J.M.; Sebti, S.M.; Hamilton, A.D. Terphenyl-based helical mimetics that disrupt the p53/HDM2 interaction. Angew. Chem. Int. Ed. Engl. 2005, 44, 2704–2707. [Google Scholar] [CrossRef] [PubMed]
- Horatscheck, A.; Wagner, S.; Ortwein, J.; Kim, B.G.; Lisurek, M.; Beligny, S.; Schutz, A.; Rademann, J. Benzoylphosphonate-based photoactive phosphopeptide mimetics for modulation of protein tyrosine phosphatases and highly specific labeling of SH2 domains. Angew. Chem. Int. Ed. Engl. 2012, 51, 9441–9447. [Google Scholar] [CrossRef] [PubMed]
- Schafer, G.; Milic, J.; Eldahshan, A.; Gotz, F.; Zuhlke, K.; Schillinger, C.; Kreuchwig, A.; Elkins, J.M.; Abdul Azeez, K.R.; Oder, A.; et al. Highly functionalized terpyridines as competitive inhibitors of AKAP-PKA interactions. Angew. Chem. Int. Ed. Engl. 2013, 52, 12187–12191. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Wells, J.A. A hot spot of binding energy in a hormone-receptor interface. Science 1995, 267, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Novotny, J.; Bruccoleri, R.E.; Saul, F.A. On the attribution of binding energy in antigen-antibody complexes McPC 603, D1.3, and HyHEL-5. Biochemistry 1989, 28, 4735–4749. [Google Scholar] [CrossRef] [PubMed]
- Pearce, K.H., Jr.; Ultsch, M.H.; Kelley, R.F.; de Vos, A.M.; Wells, J.A. Structural and mutational analysis of affinity-inert contact residues at the growth hormone-receptor interface. Biochemistry 1996, 35, 10300–10307. [Google Scholar] [CrossRef] [PubMed]
- Meinhart, A.; Alonso, J.C.; Strater, N.; Saenger, W. Crystal structure of the plasmid maintenance system epsilon/zeta: Functional mechanism of toxin zeta and inactivation by epsilon 2 zeta 2 complex formation. Proc. Natl. Acad. Sci. USA 2003, 100, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Jameson, D.M.; Seifried, S.E. Quantification of protein-protein interactions using fluorescence polarization. Methods 1999, 19, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Caldarini, M.; Sutto, L.; Camilloni, C.; Vasile, F.; Broglia, R.A.; Tiana, G. Identification of the folding inhibitors of hen-egg lysozyme: Gathering the right tools. Eur. Biophys. J. 2010, 39, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Caldarini, M.; Vasile, F.; Provasi, D.; Longhi, R.; Tiana, G.; Broglia, R.A. Identification and characterization of folding inhibitors of hen egg lysozyme: An example of a new paradigm of drug design. Proteins 2009, 74, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Beck, W.; Jung, G. Convenient reduction of S-oxides in synthetic peptides, lipopeptides and peptide libraries. Lett. Pept. Sci. 1994, 1, 31–37. [Google Scholar] [CrossRef]
- Ferrer, T.; Nicolás, E.; Giralt, E. Application of the disulfide trapping approach to explain the antiparallel assembly of dimeric rabbit uteroglobin: A preliminary study using short peptide models. Lett. Pept. Sci. 1999, 6, 165–172. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Alvarez, M.; Albericio, F. Amino acid-protecting groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramage, R.; Green, J.; Blake, A.J. An acid labile arginine derivative for peptide synthesis: NG-2,2,5,7,8-pentamethylchroman-6-sulphonyl-l-arginine. Tetrahedron 1991, 47, 6353–6370. [Google Scholar] [CrossRef]
- Vilaseca, M.; Nicolas, E.; Capdevila, F.; Giralt, E. Reduction of methionine sulfoxide with NH4I/TFA: Compatibility with peptides containing cysteine and aromatic amino acids. Tetrahedron 1998, 54, 15273–15286. [Google Scholar] [CrossRef]
- Jameson, D.M.; Croney, J.C. Fluorescence polarization: Past, present and future. Comb. Chem. High Throughput Screen. 2003, 6, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.M.; Taylor, C.W. Analysis of protein-ligand interactions by fluorescence polarization. Nat. Protoc. 2011, 6, 365–387. [Google Scholar] [CrossRef] [PubMed]
- Preidl, J.J.; Gnanapragassam, V.S.; Lisurek, M.; Saupe, J.; Horstkorte, R.; Rademann, J. Fluorescent mimetics of CMP-Neu5Ac are highly potent, cell-permeable polarization probes of eukaryotic and bacterial sialyltransferases and inhibit cellular sialylation. Angew. Chem. Int. Ed. Engl. 2014, 53, 5700–5705. [Google Scholar] [CrossRef] [PubMed]
- Huang, X. Fluorescence polarization competition assay: The range of resolvable inhibitor potency is limited by the affinity of the fluorescent ligand. J. Biomol. Screen. 2003, 8, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Spinelli, R.; Scarano, V.; Cavallo, L.; Oliva, R. COCOMAPS: A web application to analyze and visualize contacts at the interface of biomolecular complexes. Bioinformatics 2011, 27, 2915–2916. [Google Scholar] [CrossRef] [PubMed]
- Cocomaps. Available online: http://www.molnac.unisa.it/BioTools/cocomaps (accessed on 13 July 2016).
- PyMOL. Available online: www.pymol.org (accessed on 13 July 2016).
- Coin, I.; Beyermann, M.; Bienert, M. Solid-phase peptide synthesis: From standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2007, 2, 3247–3256. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Abbreviations | Peptide Sequences |
|---|---|
| I (native) | 8-TDKQFENRLNDNLEELIQ-25 |
| Ia | 18-LNDNHEELIQ-25 |
| Ib | 12-FENRLNDNHEELIQ-25 |
| Ic | 8-TRKQFENRLNDNHEELIQ-25 |
| II (native) | 43-GSGKTSLRSAIFEETQ-58 |
| IIa | 49-LRSAEFEETQ-58 |
| IIb | 45-GDTSLRSAEFEETQ-58 |
| IIc | 43-GSGDTSLRSAEFEETQ-58 |
| III (native) | 149-INSYLGTIERYETMYADD-166 |
| IIIa | 157-ERYKTMYADD-166 |
| IIIb | 153-HGTIERYKTMYADD-166 |
| IIIc | 149-INSYHGTIERYKTMYADD-166 |
| Entry | Peptide | Peptide Sequence | tR a (min) | Mass d | Binding Constant Values | ||
|---|---|---|---|---|---|---|---|
| Calculated | Found | KD (nM) e | ΔmP | ||||
| 1 | Fluo-Ia | Fluo-LNDNHEELIQ | 19.7 b | 1582.6332 | 1582.6327 | 74.5 ± 10.5 | 127 |
| 2 | Fluo-Ib | Fluo-FENRLNDNHEELIQ | 17.4 b | 2128.8883 | 2128.8901 | 89.5 ± 13.9 | 105 |
| 3 | Fluo-Ic | Fluo-TRKQFENRLNDNHEELIQ | 17.2 b | 2642.1906 | 2642.1822 | No binding | - |
| 4 | Fluo-IIa | Fluo-LRSAEFEETQ | 20.1 b | 1567.6223 | 1567.6235 | 169.5 ± 16.0 | 61 |
| 5 | Fluo-IIb | Fluo-GDTSLRSAEFEETQ | 18.1 b | 1927.7505 | 1927.7498 | 99.5 ± 12.2 | 110 |
| 6 | Fluo-IIc | Fluo-GSGDTSLRSAEFEETQ | 18.3 b | 2071.8040 | 2071.8009 | 134.0 ± 15.0 | 106 |
| 7 | Fluo-IIIa | Fluo-ERYKTMYADD | 17.4 b | 1649.6213 | 1650.2056 | No binding | - |
| 8 | Fluo-IIIb | Fluo-HGTIERYKTMYADD | 16.2 b | 2057.8334 | 2057.0183 | No binding | - |
| 9 | Fluo-IIIc | Fluo-INSYHGTIERYKTMYADD | 18.4 b | 2536.0398 | 2536.1358 | No binding | - |
| 10 | Ac-Ia | Ac-LNDNHEELIQ | 14.3 c | 1266.5961 | 1266.5998 | n/a | n/a |
| 11 | Ac-Ib | Ac-FENRLNDNHEELIQ | 14.0 c | 1812.8511 | 1812.8543 | n/a | n/a |
| 12 | Ac-Ic | Ac-TRKQFENRLNDNHEELIQ | 13.2 c | 2325.1682 | 2325.1653 | n/a | n/a |
| 13 | Ac-IIa | Ac-LRSAEFEETQ | 15.8 c | 1251.5852 | 1251.5811 | n/a | n/a |
| 14 | Ac-IIb | Ac-GDTSLRSAEFEETQ | 13.8 c | 1611.7133 | 1611.7113 | n/a | n/a |
| 15 | Ac-IIc | Ac-GSGDTSLRSAEFEETQ | 13.5 c | 1755.7668 | 1755.7641 | n/a | n/a |
| 16 | Ac-IIIa | Ac-ERYKTMYADD | 13.4 c | 1333.5841 | 1333.5891 | n/a | n/a |
| 17 | Fluo-Id | Fluo- LNANHEELIQ | 18.5 c | 1538.6434 | 1538.6474 | No binding | - |
| 18 | Fluo-Ie | Fluo-DNLNHEELIQ | 18.3 c | 1582.6332 | 1582.6327 | 175.0 ± 8.90 | 115 |
| 19 | Fluo-If | Fluo-NDNLEE | 17.5 c | 1091.3476 | 1091.3465 | 74.4 ± 8.05 | 199 |
| 20 | Fluo-Ig | Fluo-DNLEE | 17.5 c | 976.2974 | 976.2969 | 71.6 ± 9.15 | 225 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Bachiller, M.I.; Brzozowska, I.; Odolczyk, N.; Zielenkiewicz, U.; Zielenkiewicz, P.; Rademann, J. Mapping Protein–Protein Interactions of the Resistance-Related Bacterial Zeta Toxin–Epsilon Antitoxin Complex (ε2ζ2) with High Affinity Peptide Ligands Using Fluorescence Polarization. Toxins 2016, 8, 222. https://doi.org/10.3390/toxins8070222
Fernández-Bachiller MI, Brzozowska I, Odolczyk N, Zielenkiewicz U, Zielenkiewicz P, Rademann J. Mapping Protein–Protein Interactions of the Resistance-Related Bacterial Zeta Toxin–Epsilon Antitoxin Complex (ε2ζ2) with High Affinity Peptide Ligands Using Fluorescence Polarization. Toxins. 2016; 8(7):222. https://doi.org/10.3390/toxins8070222
Chicago/Turabian StyleFernández-Bachiller, María Isabel, Iwona Brzozowska, Norbert Odolczyk, Urszula Zielenkiewicz, Piotr Zielenkiewicz, and Jörg Rademann. 2016. "Mapping Protein–Protein Interactions of the Resistance-Related Bacterial Zeta Toxin–Epsilon Antitoxin Complex (ε2ζ2) with High Affinity Peptide Ligands Using Fluorescence Polarization" Toxins 8, no. 7: 222. https://doi.org/10.3390/toxins8070222
APA StyleFernández-Bachiller, M. I., Brzozowska, I., Odolczyk, N., Zielenkiewicz, U., Zielenkiewicz, P., & Rademann, J. (2016). Mapping Protein–Protein Interactions of the Resistance-Related Bacterial Zeta Toxin–Epsilon Antitoxin Complex (ε2ζ2) with High Affinity Peptide Ligands Using Fluorescence Polarization. Toxins, 8(7), 222. https://doi.org/10.3390/toxins8070222