
Role of p38alpha/beta MAP Kinase in Cell Susceptibility to Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
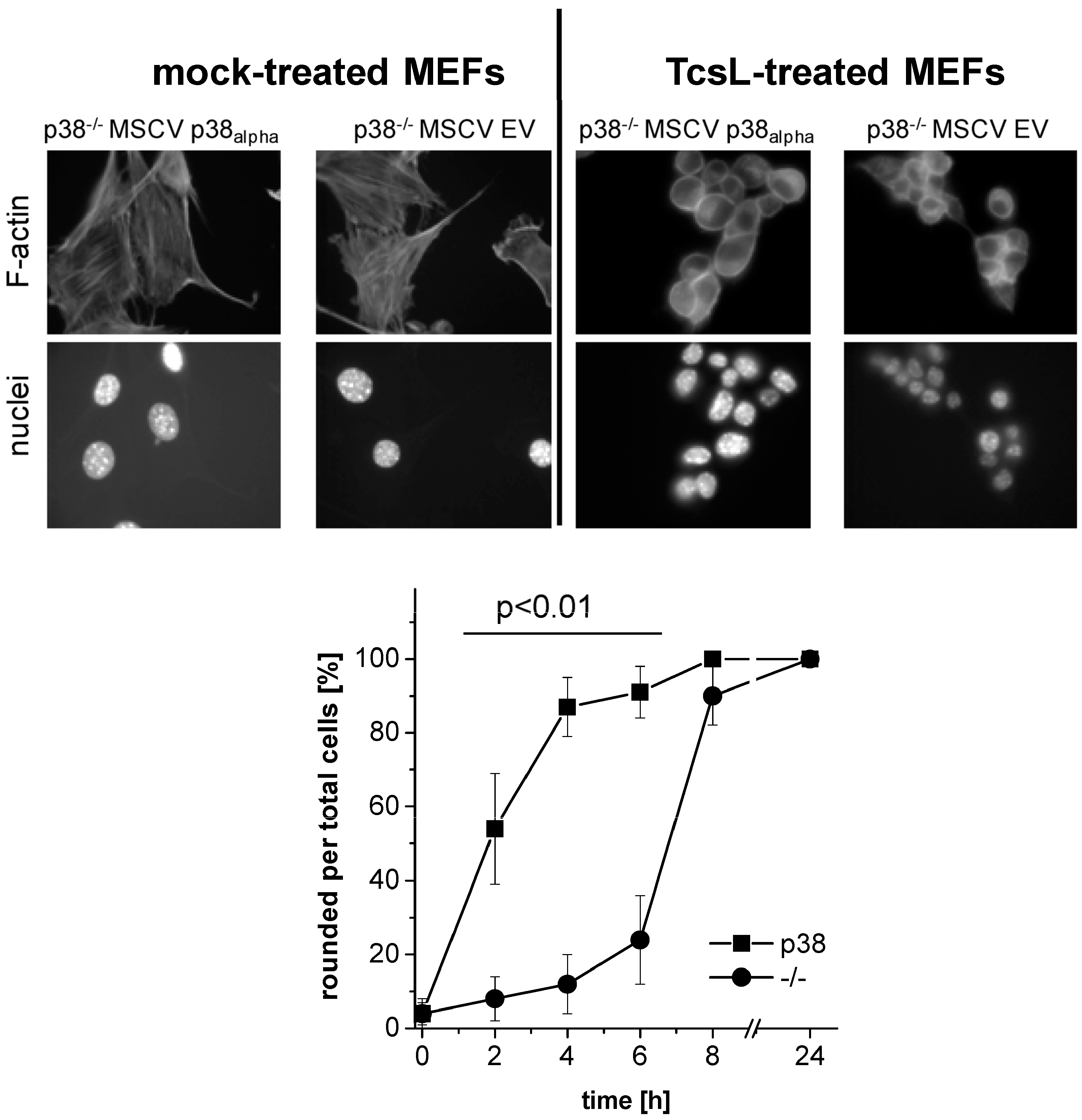
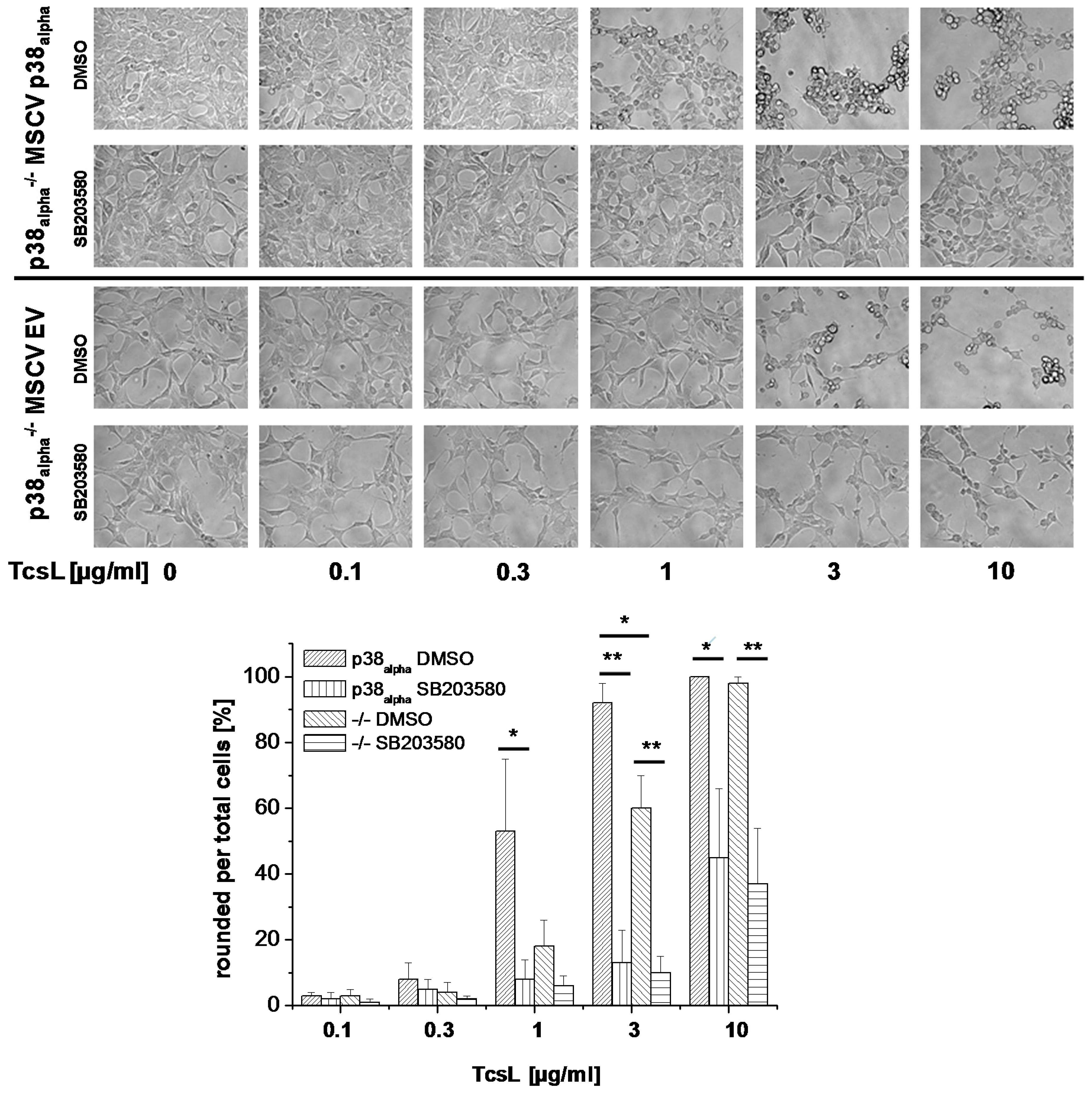
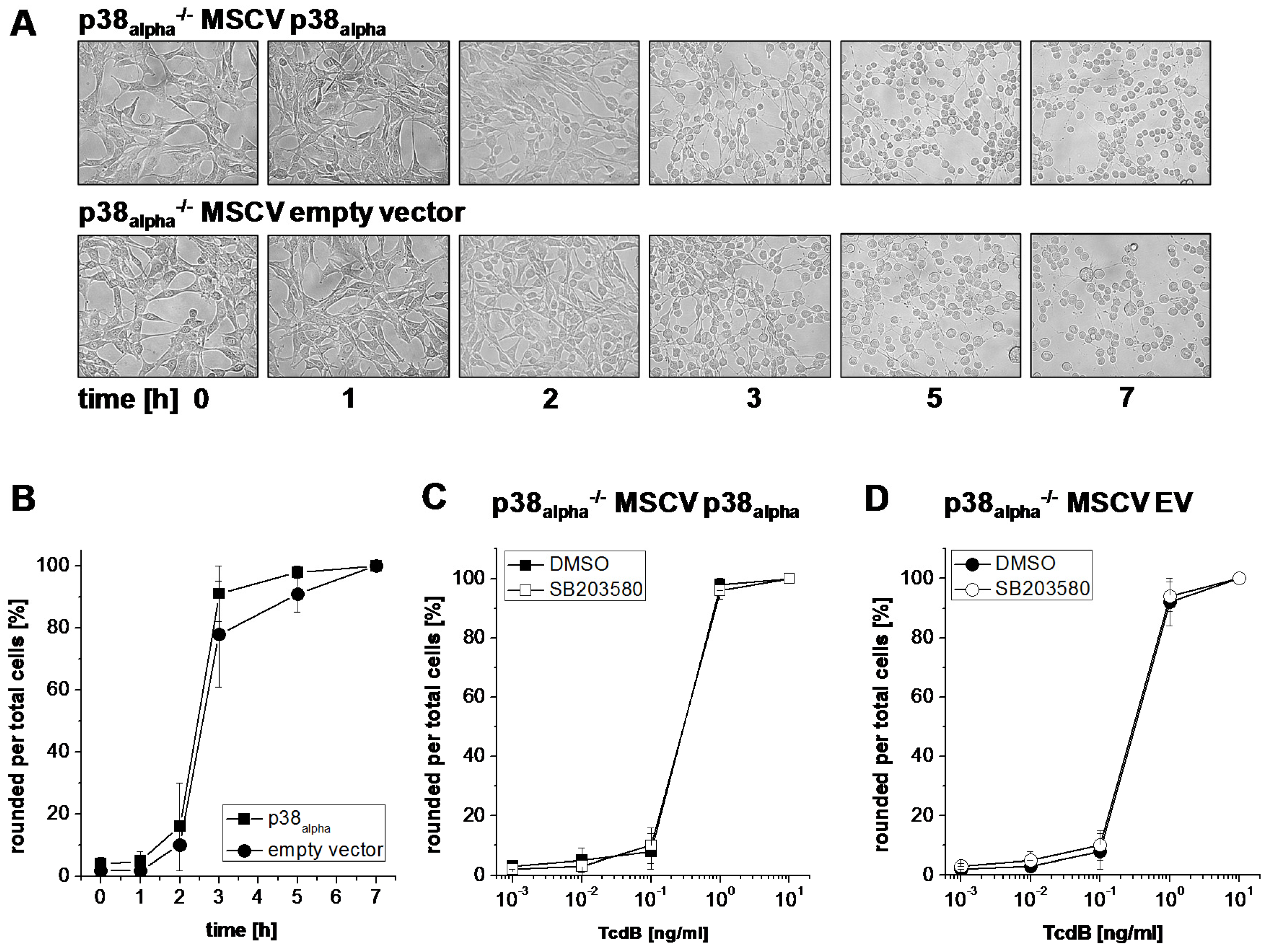
2.1. Prevention of TcsL-Induced Actin Re-Organization upon Inhibition of p38alpha/beta
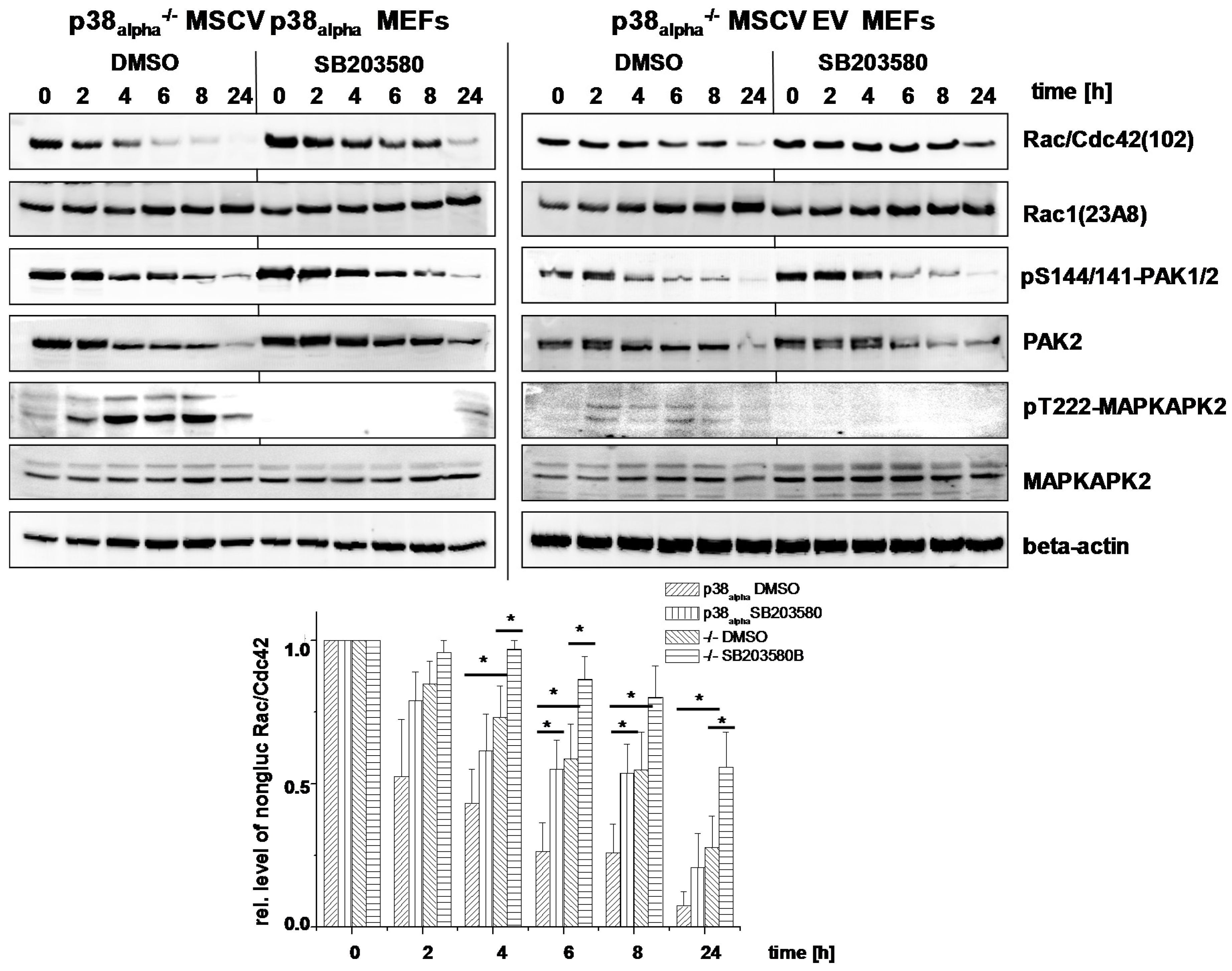
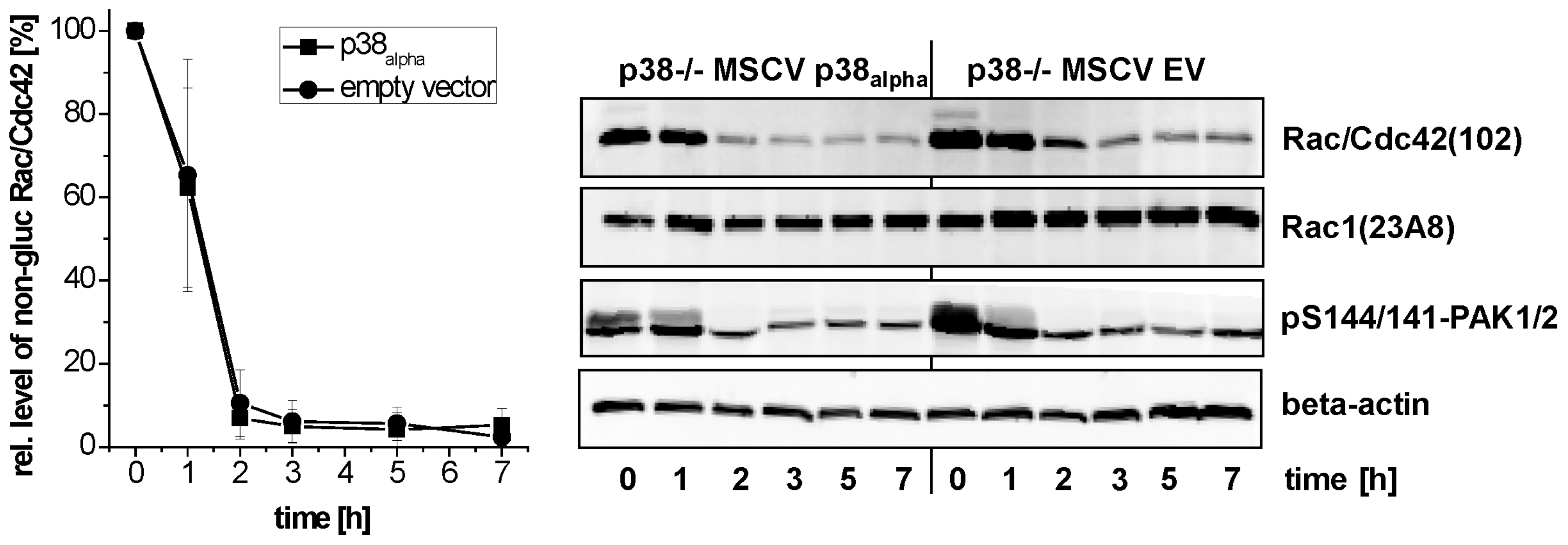
2.2. Prevention of TcsL-Induced Glucosylation of Rac/Cdc42 Subtype GTPases upon Inhibition of p38alpha/beta
2.3. SB203580 Preserves TcsL-Induced Loss of Epithelial Barrier Function
3. Discussion
4. Conclusions
- Genetic deletion of p38alpha or treatment with SB203580 protects MEFs from Rac/Cdc42 glucosylation and actin reorganization induced by TcsL (not by the related TcdB).
- Treatment with SB203580 protects epithelial monolayer from loss of epithelial barrier function induced by TcsL (not by TcdB).
- The protective effects of SB203580 treatment and of p38alpha deletion are likely based on inhibition of endocytic uptake of TcsL rather than on inhibition of the toxins’ glucosyltransferase activity.
5. Materials and Methods
5.1. Materials
5.2. Cell Culture and Preparation of Lysates
5.3. Immunoblot Analysis
5.4. Visualization of the Actin Cytoskeleton
5.5. Transepithelial Resistance of Epithelial Monolayers
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hodges, K.; Hecht, G. Bacterial infections of the small intestine. Curr. Opin. Gastroenterol. 2013, 29, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Peniche, A.G.; Savidge, T.C.; Dann, S.M. Recent insights into Clostridium difficile pathogenesis. Curr. Opin. Infect. Dis. 2013, 26, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Diab, S.S.; Songer, G.; Uzal, F.A. Clostridium difficile infection in horses: A review. Vet. Microbiol. 2013, 167, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Al-Mashat, R.R.; Taylor, D.J. Production OD diarrhoea and enteric lesions in calves by the oral inoculation of pure cultures of Clostridium sordellii. Vet. Rec. 1983, 112, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Zamboglou, C.; Genisyuerek, S.; Guttenberg, G.; Aktories, K. Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS ONE 2010, 5, e10673. [Google Scholar] [CrossRef] [PubMed]
- Jank, T.; Belyi, Y.; Aktories, K. Bacterial glycosyltransferase toxins. Cell. Microbiol. 2015, 17, 1752–1765. [Google Scholar] [CrossRef] [PubMed]
- Varela, C.C.; Haustant, G.M.; Baron, B.; England, P.; Chenal, A.; Pauillac, S.; Blondel, A.; Popoff, M.R. The Tip of the four N-Terminal alpha-helices of Clostridium sordellii lethal toxin contains the interaction site with membrane phosphatidylserine facilitating small GTPases glucosylation. Toxins (Basel) 2016, 8, E90. [Google Scholar] [CrossRef] [PubMed]
- LaFrance, M.E.; Farrow, M.A.; Chandrasekaran, R.; Sheng, J.; Rubin, D.H.; Lacy, D.B. Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 7073–7078. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Schelle, I.; Just, I. Metal Ion Activation of Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B. Toxins (Basel) 2016, 8, E109. [Google Scholar] [CrossRef] [PubMed]
- Sehr, P.; Joseph, G.; Genth, H.; Just, I.; Pick, E.; Aktories, K. Glucosylation and ADP-ribosylation of Rho proteins—Effects on nucleotide binding, GTPase activity, and effector-coupling. Biochemistry 1998, 37, 5296–5304. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Aktories, K.; Just, I. Monoglucosylation of RhoA at Threonine-37 blocks cytosol-membrane cycling. J. Biol. Chem. 1999, 274, 29050–29056. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.; Ahmadian, M.R.; Hofmann, F.; Just, I. Functional consequences of monoglucosylation of H-Ras at effector domain amino acid threonine-35. J. Biol. Chem. 1998, 273, 16134–16139. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.R.; Hofmann, F.; Wohlgemuth, S.; Herrmann, C.; Just, I. Structural consequences of mono-glucosylation of Ha-Ras by Clostridium sordellii lethal toxin. J. Mol. Biol. 2000, 301, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- May, M.; Wang, T.; Muller, M.; Genth, H. Difference in F-actin depolymerization induced by toxin B from the Clostridium difficile strain VPI 10463 and toxin B from the variant Clostridium difficile serotype F strain 1470. Toxins (Basel) 2013, 5, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Geny, B.; Grassart, A.; Manich, M.; Chicanne, G.; Payrastre, B.; Sauvonnet, N.; Popoff, M.R. Rac1 inactivation by lethal toxin from Clostridium sordellii modifies focal adhesions upstream of actin depolymerization. Cell. Microbiol. 2010, 12, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Pauillac, S.; Schelle, I.; Bouvet, P.; Bouchier, C.; Varela-Chavez, C.; Just, I.; Popoff, M.R. Haemorrhagic toxin and lethal toxin from Clostridium sordellii strain vpi9048: Molecular characterization and comparative analysis of substrate specificity of the large clostridial glucosylating toxins. Cell. Microbiol. 2014, 16, 1706–1721. [Google Scholar] [CrossRef] [PubMed]
- Lica, M.; Schulz, F.; Schelle, I.; May, M.; Just, I.; Genth, H. Difference in the biological effects of Clostridium difficile toxin B in proliferating and non-proliferating cells. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 383, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Wohlan, K.; Goy, S.; Olling, A.; Srivaratharajan, S.; Tatge, H.; Genth, H.; Gerhard, R. Pyknotic cell death induced by Clostridium difficile TcdB: Chromatin condensation and nuclear blister are induced independently of the glucosyltransferase activity. Cell. Microbiol. 2014, 16, 1678–1692. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, K.M.; Donato, G.M.; Gray, M.C.; Kolling, G.L.; Warren, C.A.; Cave, L.M.; Solga, M.D.; Lannigan, J.A.; Papin, J.A.; Hewlett, E.L. Systems analysis of the transcriptional response of human ileocecal epithelial cells to Clostridium difficile toxins and effects on cell cycle control. BMC. Syst. Biol. 2012, 6, 2. [Google Scholar]
- Arechederra, M.; Priego, N.; Vazquez-Carballo, A.; Sequera, C.; Gutierrez-Uzquiza, A.; Cerezo-Guisado, M.I.; Ortiz-Rivero, S.; Roncero, C.; Cuenda, A.; Guerrero, C.; Porras, A. p38 MAPK down-regulates fibulin 3 expression through methylation of gene regulatory sequences: Role in migration and invasion. J. Biol. Chem. 2015, 290, 4383–4397. [Google Scholar] [CrossRef] [PubMed]
- Cerezo-Guisado, M.I.; del Reino, P.; Remy, G.; Kuma, Y.; Arthur, J.S.; Gallego-Ortega, D.; Cuenda, A. Evidence of p38gamma and p38delta involvement in cell transformation processes. Carcinogenesis 2011, 32, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Geny, B.; Popoff, M.R. Activation of a c-Jun-NH2-terminal kinase pathway by the lethal toxin from Clostridium sordellii, TcsL-82, occurs independently of the toxin intrinsic enzymatic activity and facilitates small GTPase glucosylation. Cell. Microbiol. 2009, 11, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.; May, M.; Schulz, F.; Schelle, I.; Ronkina, N.; Hohenegger, M.; Fritz, G.; Just, I.; Gerhard, R.; Genth, H. Cytoprotective effect of the small GTPase RhoB expressed upon treatment of fibroblasts with the Ras-glucosylating Clostridium sordellii lethal toxin. FEBS Lett. 2012, 586, 3665–3673. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Just, I.; Genth, H. Prevention of Clostridium sordellii lethal toxin-induced apoptotic cell death by tauroursodeoxycholic acid. Biochemistry 2009, 48, 9002–9010. [Google Scholar] [CrossRef] [PubMed]
- Ronkina, N.; Menon, M.B.; Schwermann, J.; Arthur, J.S.; Legault, H.; Telliez, J.B.; Kayyali, U.S.; Nebreda, A.R.; Kotlyarov, A.; Gaestel, M. Stress induced gene expression: A direct role for MAPKAP kinases in transcriptional activation of immediate early genes. Nucleic Acids Res. 2011, 39, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Dolado, I.; Swat, A.; Ajenjo, N.; de, V.G.; Cuadrado, A.; Nebreda, A.R. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007, 11, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Halabi-Cabezon, I.; Huelsenbeck, J.; May, M.; Ladwein, M.; Rottner, K.; Just, I.; Genth, H. Prevention of the cytopathic effect induced by Clostridium difficile Toxin B by active Rac1. FEBS Lett. 2008, 582, 3751–3756. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Just, I. Functional implications of lethal toxin-catalysed glucosylation of (H/K/N)Ras and Rac1 in Clostridium sordellii-associated disease. Eur. J. Cell Biol. 2011, 90, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Huelsenbeck, J.; Hartmann, B.; Hofmann, F.; Just, I.; Gerhard, R. Cellular stability of Rho-GTPases glucosylated by Clostridium difficile toxin B. FEBS Lett. 2006, 580, 3565–3569. [Google Scholar] [CrossRef] [PubMed]
- Brandes, V.; Schelle, I.; Brinkmann, S.; Schulz, F.; Schwarz, J.; Gerhard, R.; Genth, H. Protection from Clostridium difficile toxin B-catalysed Rac1/Cdc42 glucosylation by tauroursodeoxycholic acid-induced Rac1/Cdc42 phosphorylation. Biol. Chem. 2012, 393, 77–84. [Google Scholar] [CrossRef] [PubMed]
- May, M.; Schelle, I.; Brakebusch, C.; Rottner, K.; Genth, H. Rac1-dependent recruitment of PAK2 to G phase centrosomes and their roles in the regulation of mitotic entry. Cell Cycle 2014, 13, 2210–2220. [Google Scholar] [CrossRef] [PubMed]
- Boehm, C.; Gibert, M.; Geny, B.; Popoff, M.R.; Rodriguez, P. Modification of epithelial cell barrier permeability and intercellular junctions by Clostridium sordellii lethal toxins. Cell. Microbiol. 2006, 8, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R. Bacterial factors exploit eukaryotic Rho GTPase signaling cascades to promote invasion and proliferation within their host. Small GTPases 2014, 5, e28209. [Google Scholar] [CrossRef] [PubMed]
- Zak, J.; Schneider, S.W.; Eue, I.; Ludwig, T.; Oberleithner, H. High-resistance MDCK-C7 monolayers used for measuring invasive potency of tumour cells. Pflugers Arch. 2000, 440, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Jank, T.; Giesemann, T.; Aktories, K. Clostridium difficile glucosyltransferase toxin B-essential amino acids for substrate binding. J. Biol. Chem. 2007, 282, 35222–35231. [Google Scholar] [CrossRef] [PubMed]
- Felberbaum-Corti, M.; Cavalli, V.; Gruenberg, J. Capture of the small GTPase Rab5 by GDI: Regulation by p38 MAP kinase. Methods Enzymol. 2005, 403, 367–381. [Google Scholar] [PubMed]
- Mace, G.; Miaczynska, M.; Zerial, M.; Nebreda, A.R. Phosphorylation of EEA1 by p38 MAP kinase regulates mu opioid receptor endocytosis. EMBO J. 2005, 24, 3235–3246. [Google Scholar] [CrossRef] [PubMed]
- Zwang, Y.; Yarden, Y. p38 MAP kinase mediates stress-induced internalization of EGFR: Implications for cancer chemotherapy. EMBO J. 2006, 25, 4195–4206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tanaka, T.; Sugiyama, N.; Yokoyama, S.; Kawasaki, Y.; Sakuma, T.; Ishihama, Y.; Saiki, I.; Sakurai, H. p38-Mediated phosphorylation of Eps15 endocytic adaptor protein. FEBS Lett. 2014, 588, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Kotlyarov, A.; Gaestel, M. SB202190-induced cell type-specific vacuole formation and defective autophagy do not depend on p38 MAP kinase inhibition. PLoS ONE 2011, 6, e23054. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Dhamija, S.; Kotlyarov, A.; Gaestel, M. The problem of pyridinyl imidazole class inhibitors of MAPK14/p38alpha and MAPK11/p38beta in autophagy research. Autophagy 2015, 11, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Selzer, J.; Busch, C.; Dumbach, J.; Hofmann, F.; Aktories, K.; Just, I. New method to generate enzymatically deficient Clostridium difficile toxin B as an antigen for immunization. Infect. Immun. 2000, 68, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R. Purification and characterization of Clostridium sordellii lethal toxin and cross-reactivity with Clostridium difficile cytotoxin. Infect. Immun. 1987, 55, 35–43. [Google Scholar] [PubMed]
- Ambrosino, C.; Mace, G.; Galban, S.; Fritsch, C.; Vintersten, K.; Black, E.; Gorospe, M.; Nebreda, A.R. Negative feedback regulation of MKK6 mRNA stability by p38alpha mitogen-activated protein kinase. Mol. Cell. Biol. 2003, 23, 370–381. [Google Scholar] [CrossRef] [PubMed]








© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schelle, I.; Bruening, J.; Buetepage, M.; Genth, H. Role of p38alpha/beta MAP Kinase in Cell Susceptibility to Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B. Toxins 2017, 9, 2. https://doi.org/10.3390/toxins9010002
Schelle I, Bruening J, Buetepage M, Genth H. Role of p38alpha/beta MAP Kinase in Cell Susceptibility to Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B. Toxins. 2017; 9(1):2. https://doi.org/10.3390/toxins9010002
Chicago/Turabian StyleSchelle, Ilona, Janina Bruening, Mareike Buetepage, and Harald Genth. 2017. "Role of p38alpha/beta MAP Kinase in Cell Susceptibility to Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B" Toxins 9, no. 1: 2. https://doi.org/10.3390/toxins9010002
APA StyleSchelle, I., Bruening, J., Buetepage, M., & Genth, H. (2017). Role of p38alpha/beta MAP Kinase in Cell Susceptibility to Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B. Toxins, 9(1), 2. https://doi.org/10.3390/toxins9010002