
Protection against Shiga Toxins


Abstract
:1. Introduction
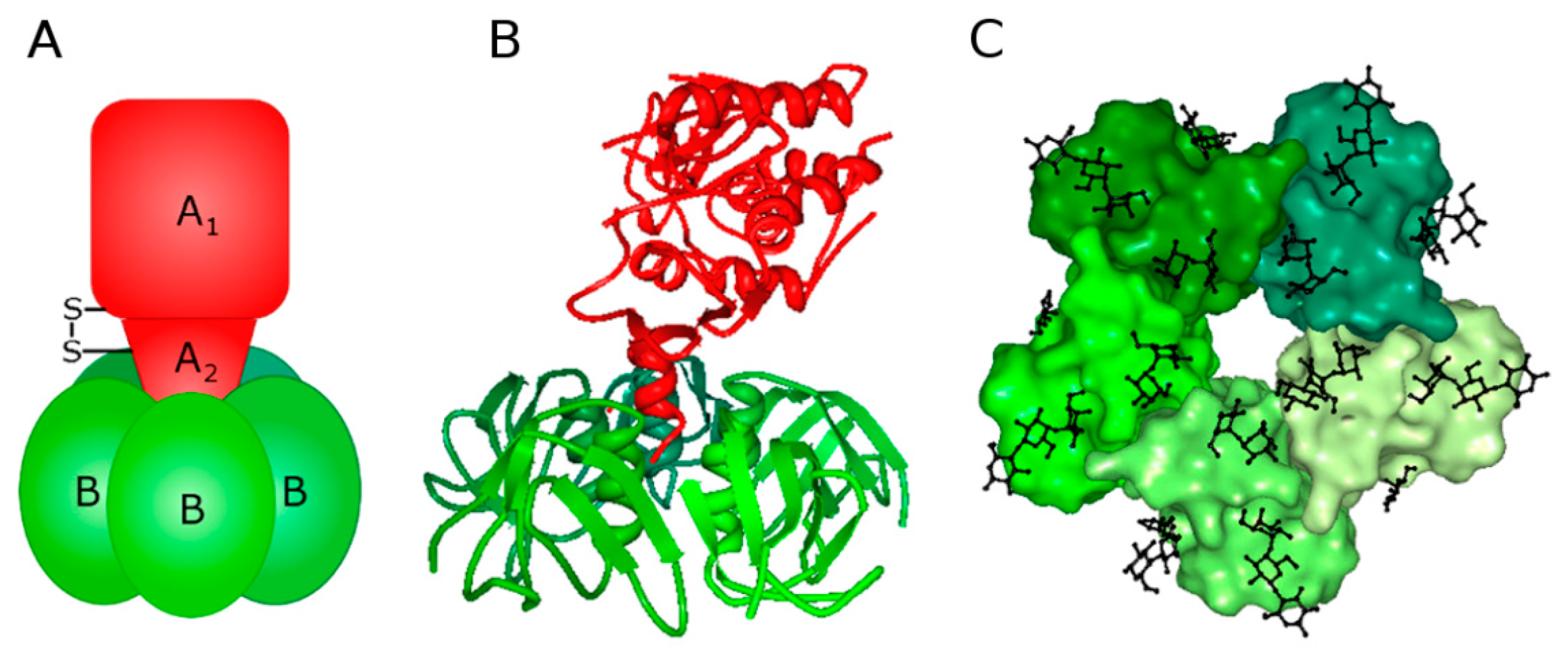
1.1. Stx Structure
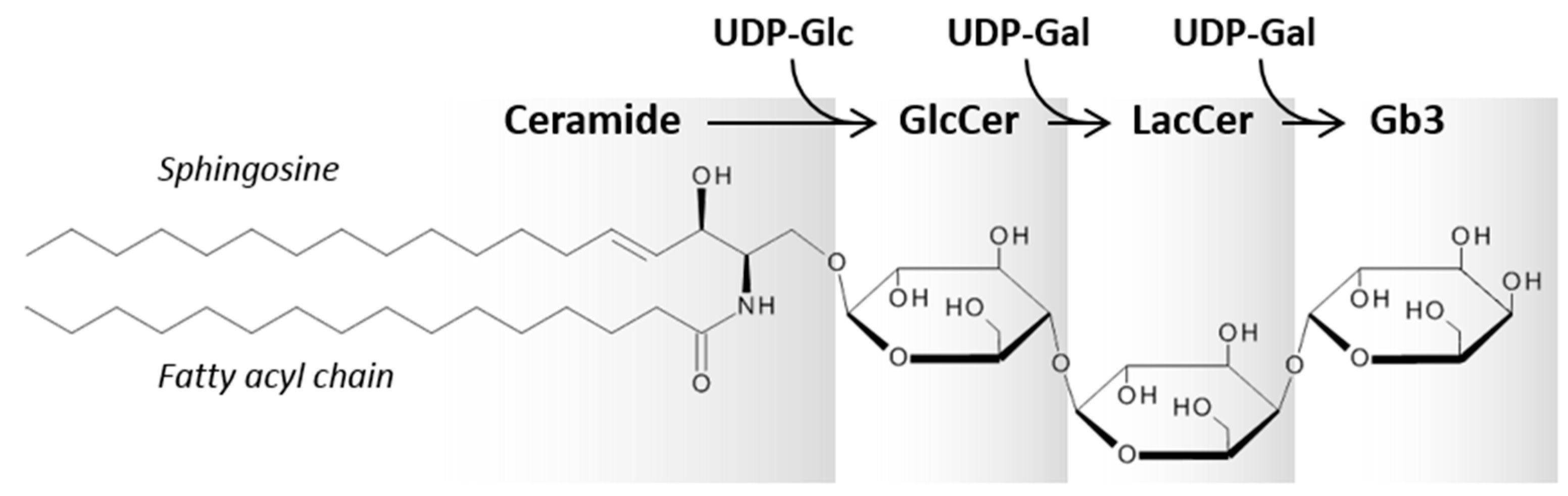
1.2. Gb3 and Its Interaction with Stx
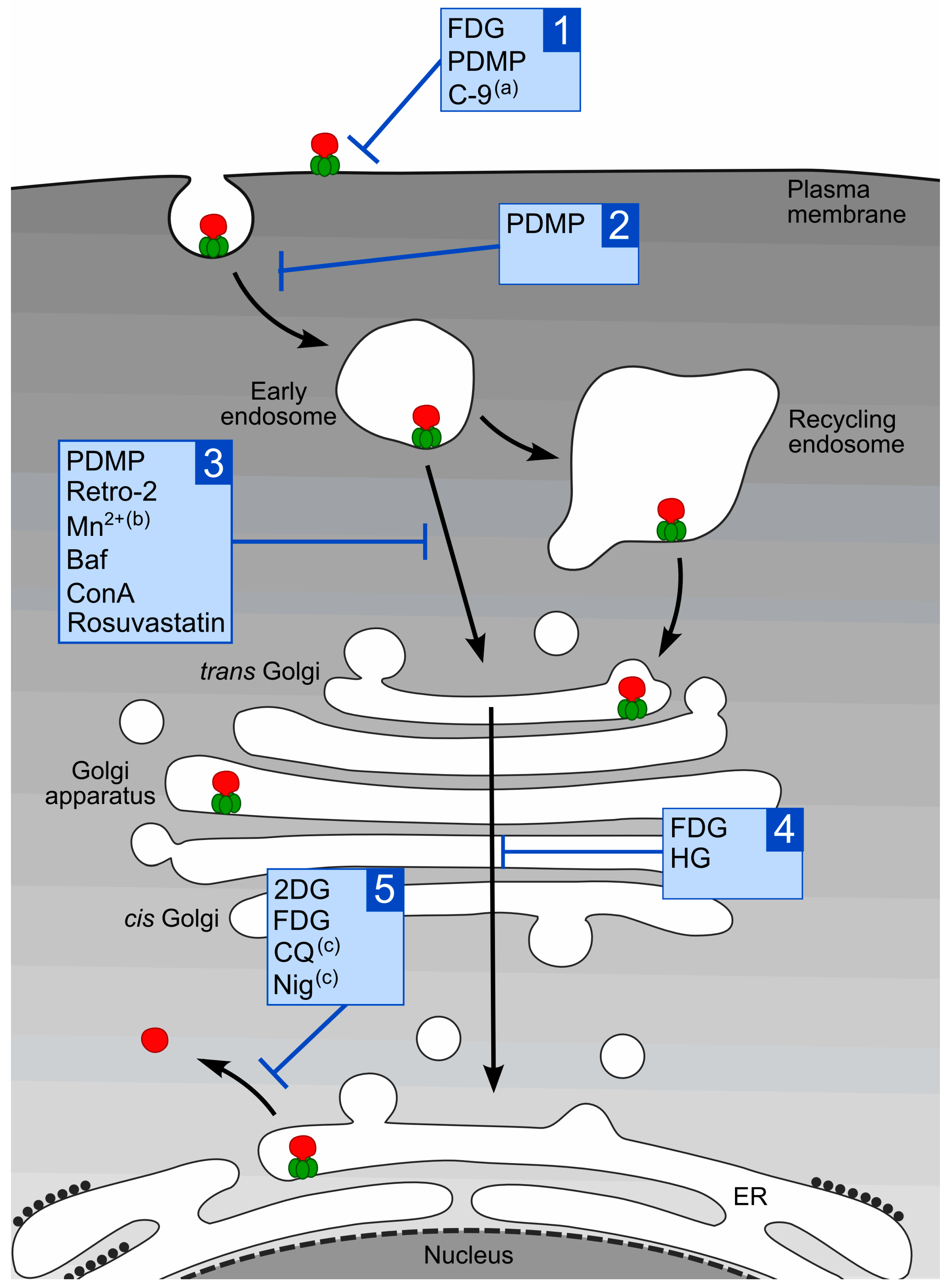
1.3. Intracellular Transport of Stx
2. Compounds that Protect Cells against Stx
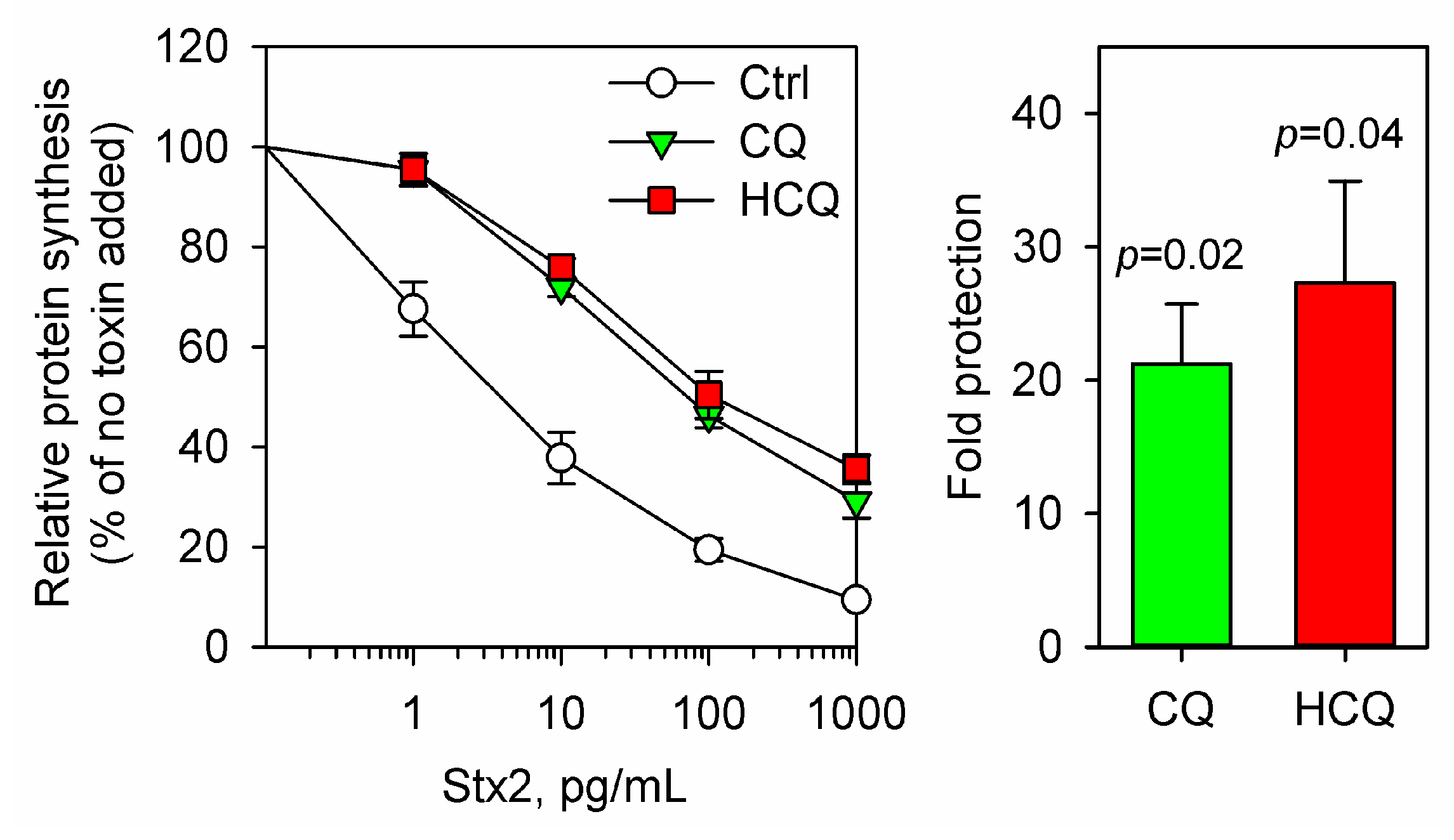
2.1. Chloroquine
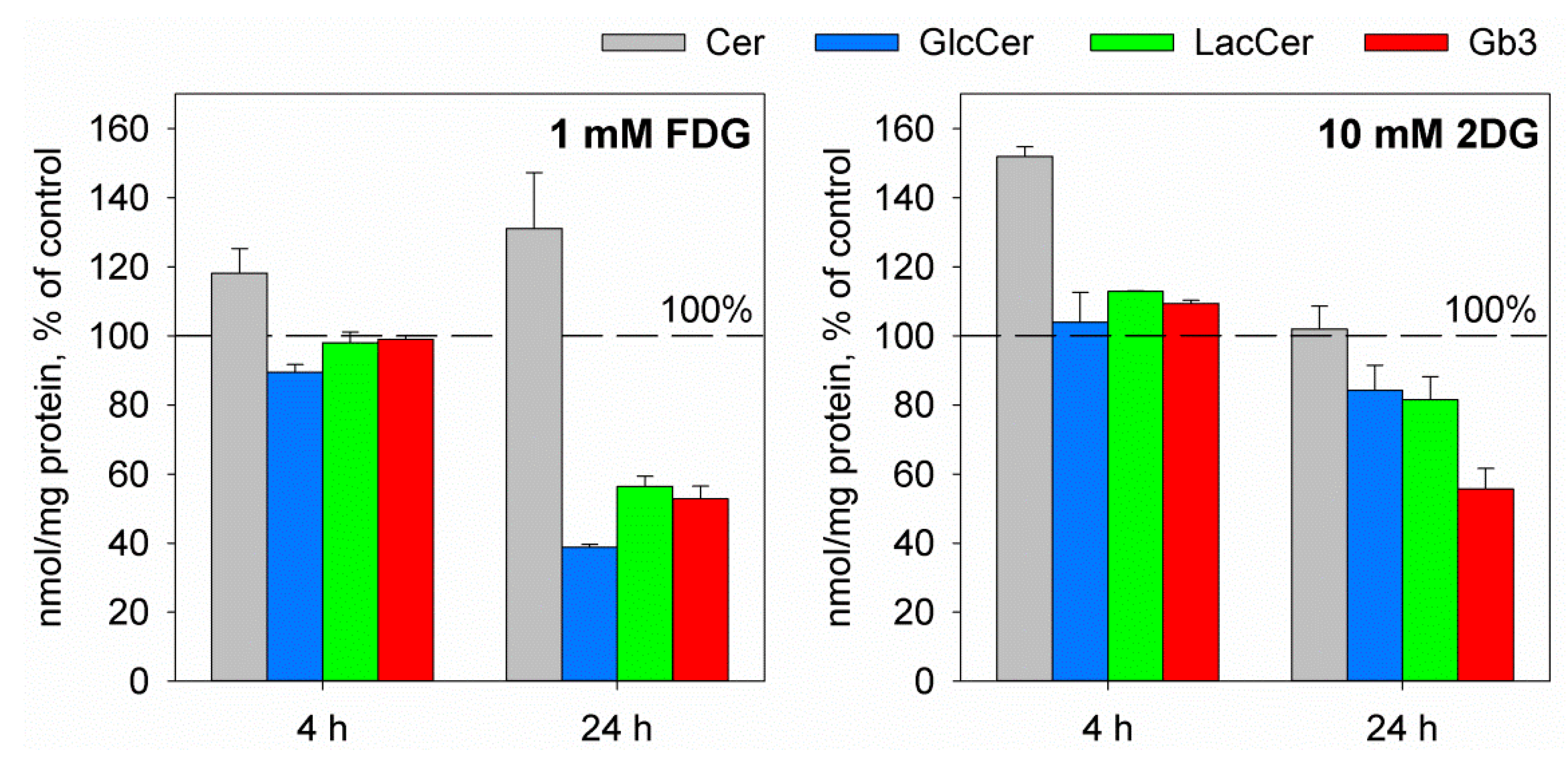
2.2. The Glucose Analogues 2-Deoxy-d-glucose and 2-Fluoro-2-deoxy-d-glucose
2.3. Retro-2 Substances
2.4. Manganese
2.5. Inhibitors of GlcCer Synthesis PDMP and C-9
2.6. HG
2.7. Statins
2.8. Furin Inhibitors
3. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| ACHN | epithelial carcinoma from renal tubular adenocarcinoma pleural metastasis |
| Baf | bafilomycin A1 |
| ConA | concanamycin A |
| cPLA2 | cytoplasmic phospholipase A2 |
| CQ | chloroquine |
| C-9 | (1R, 2R)-nonanoic acid [2-(2′,3′-dihydro-benzo [1–4]dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide-L-tartaric acid salt |
| EC50 | the concentration of the drug that gives 50% of its full inhibitory effect against the toxin |
| FDG | 2-fluoro-2-deoxy-d-glucose |
| Gal | galactose |
| Gb3 | globotriaosylceramide |
| Glc | glucose |
| GlcCer | glucosylceramide |
| GPP130 | Golgi phosphoprotein of 130 kDa |
| HBMEC | human brain microvascular endothelial cells |
| HCQ | hydroxychloroquine |
| HG | alkylglycerol 1-O-hexadecyl-sn-glycerol |
| HRTEC | human renal tubular epithelial cells |
| HUS | hemolytic-uremic syndrome |
| HuSAP | human serum amyloid component P |
| IC50 | Stx concentration needed to inhibit protein synthesis by 50% |
| LacCer | lactosylceramide |
| LPI | lysophosphatidylinositol |
| Nig | nigericin |
| PDMP | 1-phenyl-2-decanoyl-amino-3-morpholino-1- propanol |
| PET | positron emission tomography |
| Retro-2cycl | cyclic form of Retro-2 |
| STEC | Shiga toxin-producing E. coli |
| Stx | Shiga toxins (when referring to the whole family and common features) |
| StxB | B-moiety of Stx |
| Stx1 | Shiga-like toxin 1 |
| Stx2 | Shiga-like toxin 2 |
| TLC | thin layer chromatography |
| UDP | uridine diphosphate |
| [18F]FDG | FDG which contains the 18F radioisotope |
| 2DG | 2-deoxy-d-glucose |
References
- Bergan, J.; Dyve Lingelem, A.B.; Simm, R.; Skotland, T.; Sandvig, K. Shiga toxins. Toxicon 2012, 60, 1085–1107. [Google Scholar] [CrossRef] [PubMed]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L.; Burris, J.A.; Owens, J.W.; Gordon, V.M.; Wadolkowski, E.A.; O’Brien, A.D.; Samuel, J.E. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [PubMed]
- Siegler, R.L.; Obrig, T.G.; Pysher, T.J.; Tesh, V.L.; Denkers, N.D.; Taylor, F.B. Response to Shiga toxin 1 and 2 in a baboon model of hemolytic uremic syndrome. Pediatr. Nephrol. 2003, 18, 92–96. [Google Scholar] [PubMed]
- Karch, H.; Tarr, P.I.; Bielaszewska, M. Enterohaemorrhagic Escherichia coli in human medicine. Int. J. Med. Microbiol. 2005, 295, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Palermo, M.S.; Exeni, R.A.; Fernandez, G.C. Hemolytic uremic syndrome: pathogenesis and update of interventions. Expert Rev. Anti Infect. Ther. 2009, 7, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Agger, M.; Scheutz, F.; Villumsen, S.; Molbak, K.; Petersen, A.M. Antibiotic treatment of verocytotoxin-producing Escherichia coli (VTEC) infection: a systematic review and a proposal. J. Antimicrob. Chemother. 2015, 70, 2440–2446. [Google Scholar] [CrossRef] [PubMed]
- Barton Behravesh, C.; Jones, T.F.; Vugia, D.J.; Long, C.; Marcus, R.; Smith, K.; Thomas, S.; Zansky, S.; Fullerton, K.E.; Henao, O.L.; et al. Deaths associated with bacterial pathogens transmitted commonly through food: foodborne diseases active surveillance network (FoodNet), 1996–2005. J. Infect. Dis. 2011, 204, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Boisen, N.; Melton-Celsa, A.R.; Scheutz, F.; O’Brien, A.D.; Nataro, J.P. Shiga toxin 2a and Enteroaggregative Escherichia coli—a deadly combination. Gut Microbes 2015, 6, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Gould, L.H.; Demma, L.; Jones, T.F.; Hurd, S.; Vugia, D.J.; Smith, K.; Shiferaw, B.; Segler, S.; Palmer, A.; Zansky, S.; et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000–2006. Clin. Infect. Dis. 2009, 49, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.R.; Fouser, L.S.; Watkins, S.L.; Zelikovic, I.; Tarr, P.I.; Nazar-Stewart, V.; Avner, E.D. Escherichia coli O157:H7-associated hemolytic-uremic syndrome after ingestion of contaminated hamburgers. J. Pediatr. 1994, 125, 519–526. [Google Scholar] [CrossRef]
- Hughes, D.A.; Beattie, T.J.; Murphy, A.V. Haemolytic uraemic syndrome: 17 years’ experience in a Scottish paediatric renal unit. Scott. Med. J. 1991, 36, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.M.; White, R.H.; Winterborn, M.H.; Rowe, B. Haemolytic-uraemic syndrome: Clinical experience of an outbreak in the West Midlands. Br. Med. J. (Clin. Res. Ed.) 1986, 292, 1513–1516. [Google Scholar] [CrossRef]
- Bale, J.F., Jr.; Brasher, C.; Siegler, R.L. CNS manifestations of the hemolytic-uremic syndrome. Relationship to metabolic alterations and prognosis. Am. J. Dis. Child. 1980, 134, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Obata, F.; Tohyama, K.; Bonev, A.D.; Kolling, G.L.; Keepers, T.R.; Gross, L.K.; Nelson, M.T.; Sato, S.; Obrig, T.G. Shiga toxin 2 affects the central nervous system through receptor globotriaosylceramide localized to neurons. J. Infect. Dis. 2008, 198, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.D.; Fodor, E.; Vanmaele, R. Investigation of Shiga-like toxin binding to chemically synthesized oligosaccharide sequences. J. Infect. Dis. 1991, 164, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Trachtman, H.; Cnaan, A.; Christen, E.; Gibbs, K.; Zhao, S.; Acheson, D.W.; Weiss, R.; Kaskel, F.J.; Spitzer, A.; Hirschman, G.H. Effect of an oral Shiga toxin-binding agent on diarrhea-associated hemolytic uremic syndrome in children: a randomized controlled trial. JAMA 2003, 290, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Matsuoka, K.; Kita, E.; Okabe, N.; Mizuguchi, M.; Hino, K.; Miyazawa, S.; Yamasaki, C.; Aoki, J.; Takashima, S.; et al. A therapeutic agent with oriented carbohydrates for treatment of infections by Shiga toxin-producing Escherichia coli O157:H7. Proc. Natl. Acad. Sci. USA 2002, 99, 7669–7674. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, G.L.; Marcato, P.; Kitov, P.I.; Sadowska, J.; Bundle, D.R.; Armstrong, G.D. Assessment in mice of the therapeutic potential of tailored, multivalent Shiga toxin carbohydrate ligands. J. Infect. Dis. 2003, 187, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Melton-Celsa, A.R.; Carvalho, H.M.; Thuning-Roberson, C.; O’Brien, A.D. Protective efficacy and pharmacokinetics of human/mouse chimeric anti-Stx1 and anti-Stx2 antibodies in mice. Clin. Vaccine Immunol. 2015, 22, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Tani, S.; Matsumoto Yi, Y.; Takeda, T. Serum amyloid P component is the Shiga toxin 2-neutralizing factor in human blood. J. Biol. Chem. 2001, 276, 41576–41579. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.D.; Mulvey, G.L.; Marcato, P.; Griener, T.P.; Kahan, M.C.; Tennent, G.A.; Sabin, C.A.; Chart, H.; Pepys, M.B. Human serum amyloid P component protects against Escherichia coli O157:H7 Shiga toxin 2 in vivo: Therapeutic implications for hemolytic-uremic syndrome. J. Infect. Dis. 2006, 193, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Kielstein, J.T.; Beutel, G.; Fleig, S.; Steinhoff, J.; Meyer, T.N.; Hafer, C.; Kuhlmann, U.; Bramstedt, J.; Panzer, U.; Vischedyk, M.; et al. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC-HUS registry. Nephrol. Dial. Transplant. 2012, 27, 3807–3815. [Google Scholar] [CrossRef] [PubMed]
- Orth, D.; Khan, A.B.; Naim, A.; Grif, K.; Brockmeyer, J.; Karch, H.; Joannidis, M.; Clark, S.J.; Day, A.J.; Fidanzi, S.; et al. Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J. Immunol. 2009, 182, 6394–6400. [Google Scholar] [CrossRef] [PubMed]
- Melton-Celsa, A.R.; O’Brien, A.D. New therapeutic developments against Shiga toxin-producing Escherichia coli. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.E.; Boodhoo, A.; Tyrrell, G.J.; Brunton, J.L.; Read, R.J. Crystal structure of the cell-binding B oligomer of verotoxin-1 from E. coli. Nature 1992, 355, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Chernaia, M.M.; Kozlov, Y.V.; James, M.N. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 Å resolution. Nat. Struct. Biol. 1994, 1, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Jacewicz, M.; Clausen, H.; Nudelman, E.; Donohue-Rolfe, A.; Keusch, G.T. Pathogenesis of shigella diarrhea. XI. Isolation of a shigella toxin-binding glycolipid from rabbit jejunum and HeLa cells and its identification as globotriaosylceramide. J. Exp. Med. 1986, 163, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, A.A.; Brown, J.E.; Stromberg, N.; Westling-Ryd, M.; Schultz, J.E.; Karlsson, K.A. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J. Biol. Chem. 1987, 262, 1779–1785. [Google Scholar] [PubMed]
- Lingwood, C.A.; Law, H.; Richardson, S.; Petric, M.; Brunton, J.L.; De, G.S.; Karmali, M. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J. Biol. Chem. 1987, 262, 8834–8839. [Google Scholar] [PubMed]
- DeGrandis, S.; Law, H.; Brunton, J.; Gyles, C.; Lingwood, C.A. Globotetraosylceramide is recognized by the pig edema disease toxin. J. Biol. Chem. 1989, 264, 12520–12525. [Google Scholar] [PubMed]
- Ling, H.; Boodhoo, A.; Hazes, B.; Cummings, M.D.; Armstrong, G.D.; Brunton, J.L.; Read, R.J. Structure of the Shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 1998, 37, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.G.; Lingwood, C.A. Apparent cooperativity in multivalent verotoxin-globotriaosyl ceramide binding: kinetic and saturation binding studies with [125I]verotoxin. Biochim. Biophys Acta 2000, 1501, 116–124. [Google Scholar] [CrossRef]
- Bast, D.J.; Banerjee, L.; Clark, C.; Read, R.J.; Brunton, J.L. The identification of three biologically relevant globotriaosyl ceramide receptor binding sites on the Verotoxin 1 B subunit. Mol. Microbiol. 1999, 32, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Mulvey, G.; Armstrong, G.D. Cloned Shiga toxin 2 B subunit induces apoptosis in Ramos Burkitt’s lymphoma B cells. Infect. Immun. 2002, 70, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Kymre, L.; Simm, R.; Skotland, T.; Sandvig, K. Different roles of the C-terminal end of Stx1A and Stx2A for AB5 complex integrity and retrograde transport of Stx in HeLa cells. Pathog. Dis. 2015, 73, ftv083. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O’Brien, A.D.; James, M.N. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar] [CrossRef] [PubMed]
- Garred, O.; van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 1995, 270, 10817–10821. [Google Scholar] [PubMed]
- Tam, P.J.; Lingwood, C.A. Membrane cytosolic translocation of verotoxin A1 subunit in target cells. Microbiology 2007, 153, 2700–2710. [Google Scholar] [CrossRef] [PubMed]
- Zumbrun, S.D.; Hanson, L.; Sinclair, J.F.; Freedy, J.; Melton-Celsa, A.R.; Rodriguez-Canales, J.; Hanson, J.C.; O’Brien, A.D. Human intestinal tissue and cultured colonic cells contain globotriaosylceramide synthase mRNA and the alternate Shiga toxin receptor globotetraosylceramide. Infect. Immun. 2010, 78, 4488–4499. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, C.A.; Binnington, B.; Manis, A.; Branch, D.R. Globotriaosyl ceramide receptor function—where membrane structure and pathology intersect. FEBS Lett. 2010, 584, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Raa, H.; Grimmer, S.; Schwudke, D.; Bergan, J.; Walchli, S.; Skotland, T.; Shevchenko, A.; Sandvig, K. Glycosphingolipid requirements for endosome-to-Golgi transport of Shiga toxin. Traffic 2009, 10, 868–882. [Google Scholar] [CrossRef] [PubMed]
- Kiarash, A.; Boyd, B.; Lingwood, C.A. Glycosphingolipid receptor function is modified by fatty acid content. Verotoxin 1 and verotoxin 2c preferentially recognize different globotriaosyl ceramide fatty acid homologues. J. Biol. Chem. 1994, 269, 11138–11146. [Google Scholar] [PubMed]
- Pellizzari, A.; Pang, H.; Lingwood, C.A. Binding of verocytotoxin 1 to its receptor is influenced by differences in receptor fatty acid content. Biochemistry 1992, 31, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Bergan, J.; Kavaliauskiene, S.; Skotland, T. Lipid requirements for entry of protein toxins into cells. Prog. Lipid Res. 2014, 54, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kavaliauskiene, S.; Nymark, C.-M.; Bergan, J.; Simm, R.; Sylvanne, T.; Simolin, H.; Ekroos, K.; Skotland, T.; Sandvig, K. Cell density-induced changes in lipid composition and intracellular trafficking. Cell. Mol. Life Sci. 2013, 71, 1097–1116. [Google Scholar] [CrossRef] [PubMed]
- Kavaliauskiene, S.; Torgersen, M.L.; Dyve Lingelem, A.B.; Klokk, T.I.; Lintonen, T.; Simolin, H.; Ekroos, K.; Skotland, T.; Sandvig, K. Cellular effects of fluorodeoxyglucose: Global changes in the lipidome and alteration in intracellular transport. Oncotarget 2016, 7, 79885–79900. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Ryd, M.; Garred, O.; Schweda, E.; Holm, P.K.; van Deurs, B. Retrograde transport from the Golgi complex to the ER of both Shiga toxin and the nontoxic Shiga B-fragment is regulated by butyric acid and cAMP. J. Cell Biol. 1994, 126, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Bergan, J.; Skotland, T.; Sylvänne, T.; Simolin, H.; Ekroos, K.; Sandvig, K. The ether lipid precursor hexadecylglycerol causes major changes in the lipidome of HEp-2 cells. PLoS ONE 2013, 8, e75904. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.K.; Stricklett, P.K.; Schmid, D.; Kohan, D.E. Cytotoxic effect of Shiga toxin-1 on human glomerular epithelial cells. Kidney Int. 2000, 57, 2350–2359. [Google Scholar] [CrossRef] [PubMed]
- Ramegowda, B.; Samuel, J.E.; Tesh, V.L. Interaction of Shiga toxins with human brain microvascular endothelial cells: Cytokines as sensitizing agents. J. Infect. Dis. 1999, 180, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Zumbrun, S.D.; Melton-Celsa, A.R.; O’Brien, A.D. When a healthy diet turns deadly. Gut Microbes 2014, 5, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Ekroos, K.; Kavaliauskiene, S.; Bergan, J.; Kauhanen, D.; Lintonen, T.; Sandvig, K. Determining the turnover of glycosphingolipid species by stable-isotope tracer lipidomics. J. Mol. Biol. 2016, 428, 4856–4866. [Google Scholar] [CrossRef] [PubMed]
- Mahfoud, R.; Manis, A.; Lingwood, C.A. Fatty acid-dependent globotriaosyl ceramide receptor function in detergent resistant model membranes. J. Lipid Res. 2009, 50, 1744–1755. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Binnington, B.; Rog, T.; Vattulainen, I.; Grzybek, M.; Coskun, U.; Lingwood, C.A.; Simons, K. Cholesterol modulates glycolipid conformation and receptor activity. Nat. Chem. Biol. 2011, 7, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Mahfoud, R.; Manis, A.; Binnington, B.; Ackerley, C.; Lingwood, C.A. A major fraction of glycosphingolipids in model and cellular cholesterol-containing membranes is undetectable by their binding proteins. J. Biol. Chem. 2010, 285, 36049–36059. [Google Scholar] [CrossRef] [PubMed]
- Boerlin, P.; McEwen, S.A.; Boerlin-Petzold, F.; Wilson, J.B.; Johnson, R.P.; Gyles, C.L. Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J. Clin. Microbiol. 1999, 37, 497–503. [Google Scholar] [PubMed]
- Itoh, K.; Tezuka, T.; Inoue, K.; Tada, H.; Suzuki, T. Different binding property of verotoxin-1 and verotoxin-2 against their glycolipid receptor, globotriaosylceramide. Tohoku J. Exp. Med. 2001, 195, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Head, S.C.; Karmali, M.A.; Lingwood, C.A. Preparation of VT1 and VT2 hybrid toxins from their purified dissociated subunits. Evidence for B subunit modulation of a subunit function. J. Biol. Chem. 1991, 266, 3617–3621. [Google Scholar] [PubMed]
- Rutjes, N.W.; Binnington, B.A.; Smith, C.R.; Maloney, M.D.; Lingwood, C.A. Differential tissue targeting and pathogenesis of verotoxins 1 and 2 in the mouse animal model. Kidney Int. 2002, 62, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Chark, D.; Nutikka, A.; Trusevych, N.; Kuzmina, J.; Lingwood, C. Differential carbohydrate epitope recognition of globotriaosyl ceramide by verotoxins and a monoclonal antibody. Eur. J. Biochem. 2004, 271, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Proulx, F.; Lingwood, C.A. Detergent-resistant globotriaosyl ceramide may define verotoxin/glomeruli-restricted hemolytic uremic syndrome pathology. Kidney Int. 2009, 75, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Tam, P.; Mahfoud, R.; Nutikka, A.; Khine, A.A.; Binnington, B.; Paroutis, P.; Lingwood, C. Differential intracellular transport and binding of verotoxin 1 and verotoxin 2 to globotriaosylceramide-containing lipid assemblies. J. Cell. Physiol. 2008, 216, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Redler, B.; Linstedt, A.D. Shiga toxin-binding site for host cell receptor GPP130 reveals unexpected divergence in toxin-trafficking mechanisms. Mol. Biol. Cell 2013, 24, 2311–2318. [Google Scholar] [CrossRef] [PubMed]
- Selyunin, A.S.; Mukhopadhyay, S. A conserved structural motif mediates retrograde trafficking of Shiga toxin types 1 and 2. Traffic 2015, 16, 1270–1287. [Google Scholar] [CrossRef] [PubMed]
- Ergonul, Z.; Clayton, F.; Fogo, A.B.; Kohan, D.E. Shigatoxin-1 binding and receptor expression in human kidneys do not change with age. Pediatr. Nephrol. 2003, 18, 246–253. [Google Scholar] [PubMed]
- Lingwood, C.A. Verotoxin-binding in human renal sections. Nephron 1994, 66, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Boyd, B.; Lingwood, C. Verotoxin receptor glycolipid in human renal tissue. Nephron 1989, 51, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, K.; Kiyokawa, N.; Takeda, T.; Fujimoto, J. Human microvascular endothelial cells are strongly sensitive to Shiga toxins. Biochem. Biophys. Res. Commun. 1998, 251, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Iimura, M.; Kaper, J.B.; Torres, A.G.; Kagnoff, M.F. Role of Shiga toxin versus H7 flagellin in enterohaemorrhagic Escherichia coli signalling of human colon epithelium in vivo. Cell. Microbiol. 2006, 8, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Utsunomiya, I.; Taguchi, K.; Ariga, T.; Tai, T.; Ihara, Y.; Miyatake, T. Localization of verotoxin receptors in nervous system. Brain Res. 1999, 825, 183–188. [Google Scholar] [CrossRef]
- Cooling, L.L.; Walker, K.E.; Gille, T.; Koerner, T.A. Shiga toxin binds human platelets via globotriaosylceramide (Pk antigen) and a novel platelet glycosphingolipid. Infect. Immun. 1998, 66, 4355–4366. [Google Scholar] [PubMed]
- Tao, R.V.; Sweeley, C.C.; Jamieson, G.A. Sphingolipid composition of human platelets. J. Lipid Res. 1973, 14, 16–25. [Google Scholar] [PubMed]
- Steffensen, R.; Carlier, K.; Wiels, J.; Levery, S.B.; Stroud, M.; Cedergren, B.; Nilsson, S.B.; Bennett, E.P.; Jersild, C.; Clausen, H. Cloning and expression of the histo-blood group Pk UDP-galactose: Galβ1-4G1cβ1-Cer α1,4-galactosyltransferase. Molecular genetic basis of the p phenotype. J. Biol. Chem. 2000, 275, 16723–16729. [Google Scholar] [CrossRef] [PubMed]
- Mangeney, M.; Richard, Y.; Coulaud, D.; Tursz, T.; Wiels, J. CD77: An antigen of germinal center B cells entering apoptosis. Eur. J. Immunol. 1991, 21, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Engedal, N.; Skotland, T.; Torgersen, M.L.; Sandvig, K. Shiga toxin and its use in targeted cancer therapy and imaging. Microb. Biotechnol. 2011, 4, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Tokuda, N.; Numata, S.; Ito, M.; Ohta, M.; Kawamura, K.; Wiels, J.; Urano, T.; Tajima, O.; Furukawa, K.; et al. Targeted disruption of Gb3/CD77 synthase gene resulted in the complete deletion of globo-series glycosphingolipids and loss of sensitivity to verotoxins. J. Biol. Chem. 2006, 281, 10230–10235. [Google Scholar] [CrossRef] [PubMed]
- Lauvrak, S.U.; Walchli, S.; Iversen, T.G.; Slagsvold, H.H.; Torgersen, M.L.; Spilsberg, B.; Sandvig, K. Shiga toxin regulates its entry in a Syk-dependent manner. Mol. Biol. Cell 2006, 17, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, Y.U.; Mori, T.; Nakajima, H.; Katagiri, C.; Taguchi, T.; Takeda, T.; Kiyokawa, N.; Fujimoto, J. Activation of Src family kinase yes induced by Shiga toxin binding to globotriaosyl ceramide (Gb3/CD77) in low density, detergent-insoluble microdomains. J. Biol. Chem. 1999, 274, 35278–35282. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Kiyokawa, N.; Katagiri, Y.U.; Taguchi, T.; Suzuki, T.; Sekino, T.; Sato, N.; Ohmi, K.; Nakajima, H.; Takeda, T.; et al. Globotriaosyl ceramide (CD77/Gb3) in the glycolipid-enriched membrane domain participates in B-cell receptor-mediated apoptosis by regulating lyn kinase activity in human B cells. Exp. Hematol. 2000, 28, 1260–1268. [Google Scholar] [CrossRef]
- Torgersen, M.L.; Walchli, S.; Grimmer, S.; Skanland, S.S.; Sandvig, K. Protein kinase Cδ is activated by Shiga toxin and regulates its transport. J. Biol. Chem. 2007, 282, 16317–16328. [Google Scholar] [CrossRef] [PubMed]
- Walchli, S.; Skanland, S.S.; Gregers, T.F.; Lauvrak, S.U.; Torgersen, M.L.; Ying, M.; Kuroda, S.; Maturana, A.; Sandvig, K. The mitogen-activated protein kinase p38 links Shiga toxin-dependent signaling and trafficking. Mol. Biol. Cell 2008, 19, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Klokk, T.I.; Kavaliauskiene, S.; Sandvig, K. Cross-linking of glycosphingolipids at the plasma membrane: consequences for intracellular signaling and traffic. Cell. Mol. Life Sci. 2016, 73, 1301–1316. [Google Scholar] [CrossRef] [PubMed]
- Tcatchoff, L.; Andersson, S.; Utskarpen, A.; Klokk, T.I.; Skanland, S.S.; Pust, S.; Gerke, V.; Sandvig, K. Annexin A1 and A2: Roles in retrograde trafficking of Shiga toxin. PLoS ONE 2012, 7, e40429. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, H.; Kiyokawa, N.; Taguchi, T.; Matsui, J.; Katagiri, Y.U.; Okita, H.; Okuda, K.; Fujimoto, J. Shiga toxin binding to globotriaosyl ceramide induces intracellular signals that mediate cytoskeleton remodeling in human renal carcinoma-derived cells. J. Cell Sci. 2004, 117, 3911–3922. [Google Scholar] [CrossRef] [PubMed]
- Hehnly, H.; Longhini, K.M.; Chen, J.L.; Stamnes, M. Retrograde Shiga toxin trafficking is regulated by ARHGAP21 and Cdc42. Mol. Biol. Cell 2009, 20, 4303–4312. [Google Scholar] [CrossRef] [PubMed]
- Hehnly, H.; Sheff, D.; Stamnes, M. Shiga toxin facilitates its retrograde transport by modifying microtubule dynamics. Mol. Biol. Cell 2006, 17, 4379–4389. [Google Scholar] [CrossRef] [PubMed]
- Mallard, F.; Antony, C.; Tenza, D.; Salamero, J.; Goud, B.; Johannes, L. Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of Shiga toxin B-fragment transport. J. Cell Biol. 1998, 143, 973–990. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Olsnes, S.; Brown, J.E.; Petersen, O.W.; van Deurs, B. Endocytosis from coated pits of Shiga toxin: a glycolipid-binding protein from Shigella dysenteriae 1. J. Cell Biol. 1989, 108, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Donta, S.T.; Tomicic, T.K.; Donohue-Rolfe, A. Inhibition of Shiga-like toxins by brefeldin A. J. Infect. Dis. 1995, 171, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Prydz, K.; Ryd, M.; van Deurs, B. Endocytosis and intracellular transport of the glycolipid-binding ligand Shiga toxin in polarized MDCK cells. J. Cell Biol. 1991, 113, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Garred, O.; Dubinina, E.; Polesskaya, A.; Olsnes, S.; Kozlov, J.; Sandvig, K. Role of the disulfide bond in Shiga toxin A-chain for toxin entry into cells. J. Biol. Chem. 1997, 272, 11414–11419. [Google Scholar] [PubMed]
- Garred, O.; Dubinina, E.; Holm, P.K.; Olsnes, S.; van Deurs, B.; Kozlov, J.V.; Sandvig, K. Role of processing and intracellular transport for optimal toxicity of Shiga toxin and toxin mutants. Exp. Cell Res. 1995, 218, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Romer, W. Shiga toxin—From cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Koo, S.; Jeong, D.G.; Tesh, V.L. Shiga toxins as multi-functional proteins: Induction of host cellular stress responses, role in pathogenesis and therapeutic applications. Toxins (Basel) 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Dyve Lingelem, A.B.; Bergan, J.; Sandvig, K. Inhibitors of intravesicular acidification protect against Shiga toxin in a pH-independent manner. Traffic 2012, 13, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Kavaliauskiene, S.; Skotland, T.; Sylvanne, T.; Simolin, H.; Klokk, T.I.; Torgersen, M.L.; Lingelem, A.B.; Simm, R.; Ekroos, K.; Sandvig, K. Novel actions of 2-deoxy-d-glucose: protection against Shiga toxins and changes in cellular lipids. Biochem. J. 2015, 470, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Stechmann, B.; Bai, S.K.; Gobbo, E.; Lopez, R.; Merer, G.; Pinchard, S.; Panigai, L.; Tenza, D.; Raposo, G.; Beaumelle, B.; et al. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 2010, 141, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Secher, T.; Shima, A.; Hinsinger, K.; Cintrat, J.C.; Johannes, L.; Barbier, J.; Gillet, D.; Oswald, E. Retrograde trafficking inhibitor of Shiga toxins reduces morbidity and mortality of mice infected with enterohemorrhagic Esherichia coli. Antimicrob. Agents Chemother. 2015, 59, 5010–5013. [Google Scholar] [CrossRef]
- Tewari, R.; Jarvela, T.; Linstedt, A.D. Manganese induces oligomerization to promote down-regulation of the intracellular trafficking receptor used by Shiga toxin. Mol. Biol. Cell 2014, 25, 3049–3058. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Linstedt, A.D. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science 2012, 335, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Gaston, M.A.; Pellino, C.A.; Weiss, A.A. Failure of manganese to protect from Shiga toxin. PLoS ONE 2013, 8, e69823. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Brown, J.E. Ionic requirements for entry of Shiga toxin from Shigella dysenteriae 1 into cells. Infect. Immun. 1987, 55, 298–303. [Google Scholar] [PubMed]
- Silberstein, C.; Copeland, D.P.; Chiang, W.; Repetto, H.A.; Ibarra, C. A glucosylceramide synthase inhibitor prevents the cytotoxic effects of Shiga toxin-2 on human renal tubular epithelial cells. J. Epithel. Biol. Pharmacol. 2008, 1, 71–75. [Google Scholar] [CrossRef]
- Silberstein, C.; Lucero, M.S.; Zotta, E.; Copeland, D.P.; Lingyun, L.; Repetto, H.A.; Ibarra, C. A glucosylceramide synthase inhibitor protects rats against the cytotoxic effects of Shiga toxin 2. Pediatr. Res. 2011, 69, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.M.; Sacerdoti, F.; Jancic, C.; Repetto, H.A.; Paton, A.W.; Paton, J.C.; Ibarra, C. Action of Shiga toxin type-2 and subtilase cytotoxin on human microvascular endothelial cells. PLoS ONE 2013, 8, e70431. [Google Scholar] [CrossRef] [PubMed]
- Bergan, J.; Skotland, T.; Lingelem, A.B.; Simm, R.; Spilsberg, B.; Lindback, T.; Sylvanne, T.; Simolin, H.; Ekroos, K.; Sandvig, K. The ether lipid precursor hexadecylglycerol protects against Shiga toxins. Cell. Mol. Life Sci. 2014, 71, 4285–4300. [Google Scholar] [CrossRef] [PubMed]
- Binnington, B.; Nguyen, L.; Kamani, M.; Hossain, D.; Marks, D.L.; Budani, M.; Lingwood, C.A. Inhibition of Rab prenylation by statins induces cellular glycosphingolipid remodeling. Glycobiology 2016, 26, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Becker, G.L.; Lu, Y.; Hardes, K.; Strehlow, B.; Levesque, C.; Lindberg, I.; Sandvig, K.; Bakowsky, U.; Day, R.; Garten, W.; et al. Highly potent inhibitors of proprotein convertase furin as potential drugs for treatment of infectious diseases. J. Biol. Chem. 2012, 287, 21992–22003. [Google Scholar] [CrossRef] [PubMed]
- Hardes, K.; Becker, G.L.; Lu, Y.; Dahms, S.O.; Kohler, S.; Beyer, W.; Sandvig, K.; Yamamoto, H.; Lindberg, I.; Walz, L.; et al. Novel Furin inhibitors with potent anti-infectious activity. ChemMedChem 2015, 10, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Meshnick, S.R.; Dobson, M.J. The history of antimalarial drugs. In Antimalarial Chemotherapy: Mechanisms of Action, Resistance, and New Directions in Drug Discovery; Rosenthal, P.J., Ed.; Humana Press: Totowa, NJ, USA, 2001; pp. 15–25. [Google Scholar]
- Kitchen, L.W.; Vaughn, D.W.; Skillman, D.R. Role of US military research programs in the development of US Food and Drug Administration—Approved antimalarial drugs. Clin. Infect. Dis. 2006, 43, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Al-Bari, M.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, S. Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies. Eur. J. Pharmacol. 2016, 771, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Keeling, D.J.; Herslof, M.; Ryberg, B.; Sjogren, S.; Solvell, L. Vacuolar H(+)-ATPases. Targets for drug discovery? Ann. N. Y. Acad. Sci. 1997, 834, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Parra, K.J. Vacuolar ATPase: A model proton pump for antifungal drug discovery. In Antimicrobial Drug Discovery: Emerging Strategies; Tegos, G., Mylonakis, E., Eds.; CAB International: Accra, Ghana, 2012; pp. 89–100. [Google Scholar]
- Moreau, D.; Kumar, P.; Wang, S.C.; Chaumet, A.; Chew, S.Y.; Chevalley, H.; Bard, F. Genome-wide RNAi screens identify genes required for Ricin and PE intoxications. Dev. Cell 2011, 21, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Haslam, D.B. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERDj3. Infect. Immun. 2005, 73, 2524–2532. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Menchaca, A.A.; Navarro-Polanco, R.A.; Ferrer-Villada, T.; Rupp, J.; Sachse, F.B.; Tristani-Firouzi, M.; Sanchez-Chapula, J.A. The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc. Natl. Acad. Sci. USA 2008, 105, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Orlik, F.; Schiffler, B.; Benz, R. Anthrax toxin protective antigen: inhibition of channel function by chloroquine and related compounds and study of binding kinetics using the current noise analysis. Biophys J. 2005, 88, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Bachmeyer, C.; Benz, R.; Barth, H.; Aktories, K.; Gilbert, M.; Popoff, M.R. Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes and Vero cells: inhibition of channel function by chloroquine and related compounds in vitro and intoxification in vivo. FASEB J. 2001, 15, 1658–1660. [Google Scholar] [CrossRef] [PubMed]
- Browning, D.J. Pharmacology of chloroquine and hydroxychloroquine. In Hydroxychloroquine and Chloroquine Retinopathy; Springer: New York, NY, USA, 2014; pp. 35–63. [Google Scholar]
- Molina, D.K. Postmortem hydroxychloroquine concentrations in nontoxic cases. Am. J. Forensic Med. Pathol. 2012, 33, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Brown, J. Effects of 2-deoxyglucose on carbohydrate metablism: review of the literature and studies in the rat. Metabolism 1962, 11, 1098–1112. [Google Scholar] [PubMed]
- Wick, A.N.; Drury, D.R.; Nakada, H.I.; Wolfe, J.B. Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 1957, 224, 963–969. [Google Scholar] [PubMed]
- Cramer, F.B.; Woodward, G.E. 2-Desoxy-d-glucose as an antagonist of glucose in yeast fermentation. J. Frankl. Inst. 1952, 253, 354–360. [Google Scholar] [CrossRef]
- Sols, A.; Crane, R.K. Substrate specificity of brain hexokinase. J. Biol. Chem. 1954, 210, 581–595. [Google Scholar] [PubMed]
- Chen, W.; Gueron, M. The inhibition of bovine heart hexokinase by 2-deoxy-d-glucose-6-phosphate: characterization by 31P NMR and metabolic implications. Biochimie 1992, 74, 867–873. [Google Scholar] [CrossRef]
- Datema, R.; Schwarz, R.T. Interference with glycosylation of glycoproteins. Inhibition of formation of lipid-linked oligosaccharides in vivo. Biochem. J. 1979, 184, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Desselle, A.; Chaumette, T.; Gaugler, M.H.; Cochonneau, D.; Fleurence, J.; Dubois, N.; Hulin, P.; Aubry, J.; Birkle, S.; Paris, F. Anti-Gb3 monoclonal antibody inhibits angiogenesis and tumor development. PLoS ONE 2012, 7, e45423. [Google Scholar] [CrossRef] [PubMed]
- Watowich, S.S.; Morimoto, R.I. Complex regulation of heat shock- and glucose-responsive genes in human cells. Mol. Cell. Biol. 1988, 8, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Shinjo, S.; Mizotani, Y.; Tashiro, E.; Imoto, M. Comparative analysis of the expression patterns of UPR-target genes caused by UPR-inducing compounds. Biosci. Biotechnol. Biochem. 2013, 77, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Furukawa, K.; Nakayama, K.I. A novel, promoter-based, target-specific assay identifies 2-deoxy-d-glucose as an inhibitor of globotriaosylceramide biosynthesis. FEBS J. 2009, 276, 5191–5202. [Google Scholar] [CrossRef] [PubMed]
- Kurtoglu, M.; Maher, J.C.; Lampidis, T.J. Differential toxic mechanisms of 2-deoxy-d-glucose versus 2-fluorodeoxy-d-glucose in hypoxic and normoxic tumor cells. Antioxid. Redox Signal. 2007, 9, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Lampidis, T.J.; Kurtoglu, M.; Maher, J.C.; Liu, H.; Krishan, A.; Sheft, V.; Szymanski, S.; Fokt, I.; Rudnicki, W.R.; Ginalski, K.; et al. Efficacy of 2-halogen substituted d-glucose analogs in blocking glycolysis and killing “hypoxic tumor cells”. Cancer Chemother. Pharmacol. 2006, 58, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Datema, R.; Schwarz, R.T.; Jankowski, A.W. Fluoroglucose-inhibition of protein glycosylation in vivo. Inhibition of mannose and glucose incorporation into lipid-linked oligosaccharides. Eur. J. Biochem. 1980, 109, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.F.; Biely, P.; Kratky, Z.; Schwarz, R.T. Metabolism of 2-deoxy-2-fluoro-d-[3H]glucose and 2-deoxy-2-fluoro-d-[3H]mannose in yeast and chick-embryo cells. Eur. J. Biochem. 1978, 87, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Hoh, C.K. Clinical use of FDG-PET. Nucl. Med. Biol. 2007, 34, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Kelloff, G.J.; Hoffman, J.M.; Johnson, B.; Scher, H.I.; Siegel, B.A.; Cheng, E.Y.; Cheson, B.D.; O’Shaughnessy, J.; Guyton, K.Z.; Mankoff, D.A.; et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin. Cancer. Res. 2005, 11, 2785–2808. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Barredo, J.C.; Merchan, J.R.; Lampidis, T.J. Endoplasmic reticulum stress induced by 2-deoxyglucose but not glucose starvation activates AMPK through CAMKKβ leading to autophagy. Biochem. Pharmacol. 2013, 85, 1463–1477. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Kurtoglu, M.; Liu, H.; Wangpaichitr, M.; You, M.; Liu, X.; Savaraj, N.; Lampidis, T.J. 2-Deoxy-d-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 2011, 67, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Biswas, C.; Ostrovsky, O.; Makarewich, C.A.; Wanderling, S.; Gidalevitz, T.; Argon, Y. The peptide-binding activity of GRP94 is regulated by calcium. Biochem. J. 2007, 405, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Hendershot, L.M. The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. 2004, 71, 289–297. [Google Scholar] [PubMed]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Saenz, J.B.; Doggett, T.A.; Haslam, D.B. Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect. Immun. 2007, 75, 4552–4561. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Kahn, J.N.; Tumer, N.E.; Pang, Y.P. Chemical structure of Retro-2, a compound that protects cells against ribosome-inactivating proteins. Sci. Rep. 2012, 2, 631. [Google Scholar] [CrossRef] [PubMed]
- Noel, R.; Gupta, N.; Pons, V.; Goudet, A.; Garcia-Castillo, M.D.; Michau, A.; Martinez, J.; Buisson, D.A.; Johannes, L.; Gillet, D.; et al. N-methyldihydroquinazolinone derivatives of Retro-2 with enhanced efficacy against Shiga toxin. J. Med. Chem. 2013, 56, 3404–3413. [Google Scholar] [CrossRef] [PubMed]
- Carney, D.W.; Nelson, C.D.; Ferris, B.D.; Stevens, J.P.; Lipovsky, A.; Kazakov, T.; DiMaio, D.; Atwood, W.J.; Sello, J.K. Structural optimization of a retrograde trafficking inhibitor that protects cells from infections by human polyoma- and papillomaviruses. Bioorg. Med. Chem. 2014, 22, 4836–4847. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Pons, V.; Noel, R.; Buisson, D.A.; Michau, A.; Johannes, L.; Gillet, D.; Barbier, J.; Cintrat, J.C. (S)-N-Methyldihydroquinazolinones are the active enantiomers of Retro-2 derived compounds against toxins. ACS Med. Chem. Lett. 2014, 5, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Mellmann, A.; Zhang, W.; Kock, R.; Fruth, A.; Bauwens, A.; Peters, G.; Karch, H. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: a microbiological study. Lancet Infect. Dis. 2011, 11, 671–676. [Google Scholar] [CrossRef]
- Gupta, N.; Noel, R.; Goudet, A.; Hinsinger, K.; Michau, A.; Pons, V.; Abdelkafi, H.; Secher, T.; Shima, A.; Shtanko, O.; et al. Inhibitors of retrograde trafficking active against ricin and Shiga toxins also protect cells from several viruses, Leishmania and Chlamydiales. Chem. Biol. Interact. 2016. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Erikson, K.M.; Herrero Hernandez, E.; Tjalkens, R. Manganese and its role in Parkinson’s disease: from transport to neuropathology. Neuromol. Med. 2009, 11, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Racette, B.A.; Aschner, M.; Guilarte, T.R.; Dydak, U.; Criswell, S.R.; Zheng, W. Pathophysiology of manganese-associated neurotoxicity. Neurotoxicology 2012, 33, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Vunnam, R.R.; Radin, N.S. Analogs of ceramide that inhibit glucocerebroside synthetase in mouse brain. Chem. Phys. Lipids 1980, 26, 265–278. [Google Scholar] [CrossRef]
- Jmoudiak, M.; Futerman, A.H. Gaucher disease: pathological mechanisms and modern management. Br. J. Haematol. 2005, 129, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Barbour, S.; Edidin, M.; Felding-Habermann, B.; Taylor-Norton, J.; Radin, N.S.; Fenderson, B.A. Glycolipid depletion using a ceramide analogue (PDMP) alters growth, adhesion, and membrane lipid organization in human A431 cells. J. Cell Physiol. 1992, 150, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Arab, S.; Lingwood, C.A. Intracellular targeting of the endoplasmic reticulum/nuclear envelope by retrograde transport may determine cell hypersensitivity to verotoxin via globotriaosyl ceramide fatty acid isoform traffic. J. Cell. Physiol. 1998, 177, 646–660. [Google Scholar] [CrossRef]
- Sandvig, K.; Garred, O.; van Helvoort, A.; van Meer, G.; van Deurs, B. Importance of glycolipid synthesis for butyric acid-induced sensitization to Shiga toxin and intracellular sorting of toxin in A431 cells. Mol. Biol. Cell 1996, 7, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Abe, A.; Shayman, J.A. Improved inhibitors of glucosylceramide synthase. J. Biol. Chem. 1999, 274, 14662–14669. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Dastgheib, S.; Girzadas, M.A.; O’Donnell, P.H.; Westervelt, C.W.; Li, Z.; Inokuchi, J.; Basu, S. Hydrophobic nature of mammalian ceramide glycanases: purified from rabbit and rat mammary tissues. Acta Biochim. Pol. 1998, 45, 327–342. [Google Scholar] [PubMed]
- Abe, A.; Gregory, S.; Lee, L.; Killen, P.D.; Brady, R.O.; Kulkarni, A.; Shayman, J.A. Reduction of globotriaosylceramide in Fabry disease mice by substrate deprivation. J. Clin. Invest. 2000, 105, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.M. Eliglustat: first global approval. Drugs 2014, 74, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Belmatoug, N.; Di Rocco, M.; Fraga, C.; Giraldo, P.; Hughes, D.; Lukina, E.; Maison-Blanche, P.; Merkel, M.; Niederau, C.; Plckinger, U.; et al. Management and monitoring recommendations for the use of eliglustat in adults with type 1 Gaucher disease in Europe. Eur. J. Intern. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lukina, E.; Watman, N.; Arreguin, E.A.; Banikazemi, M.; Dragosky, M.; Iastrebner, M.; Rosenbaum, H.; Phillips, M.; Pastores, G.M.; Rosenthal, D.I.; et al. A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood 2010, 116, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Das, A.K.; Holmes, R.D.; Wilson, G.N.; Hajra, A.K. Dietary ether lipid incorporation into tissue plasmalogens of humans and rodents. Lipids 1992, 27, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Iannitti, T.; Palmieri, B. An update on the therapeutic role of alkylglycerols. Mar. Drugs 2010, 8, 2267–2300. [Google Scholar] [CrossRef] [PubMed]
- Deniau, A.L.; Mosset, P.; Pedrono, F.; Mitre, R.; Le Bot, D.; Legrand, A.B. Multiple beneficial health effects of natural alkylglycerols from shark liver oil. Mar. Drugs 2010, 8, 2175–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matusewicz, L.; Meissner, J.; Toporkiewicz, M.; Sikorski, A.F. The effect of statins on cancer cells—Review. Tumour Biol. 2015, 36, 4889–4904. [Google Scholar] [CrossRef] [PubMed]
- Correale, M.; Abruzzese, S.; Greco, C.A.; Concilio, M.; Di Biase, M.; Brunetti, N.D. Statins in heart failure. Curr. Vasc. Pharmacol. 2012, 69, 232. [Google Scholar] [CrossRef]
- Paliani, U.; Ricci, S. The role of statins in stroke. Intern. Emerg. Med. 2012, 7, 305–311. [Google Scholar] [CrossRef] [PubMed]
- US Preventive Services Task Force; Bibbins-Domingo, K.; Grossman, D.C.; Curry, S.J.; Davidson, K.W.; Epling, J.W.; Garcia, F.A.; Gillman, M.W.; Kemper, A.R.; Krist, A.H.; et al. Statin use for the primary prevention of cardiovascular disease in adults: US preventive services task force recommendation statement. JAMA 2016, 316, 1997–2007. [Google Scholar] [PubMed]
- Oda, T.; Wu, H.C. Effect of lovastatin on the cytotoxicity of ricin, modeccin, Pseudomonas toxin, and diphtheria toxin in brefeldin A-sensitive and -resistant cell lines. Exp. Cell Res. 1994, 212, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.Q.; Ali, B.R.; Ramalho, J.S.; Godfrey, R.F.; Barral, D.C.; Hume, A.N.; Seabra, M.C. Membrane targeting of Rab GTPases is influenced by the prenylation motif. Mol. Biol. Cell 2003, 14, 1882–1899. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Aivazian, D. Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 2004, 5, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Holstein, S.A.; Knapp, H.R.; Clamon, G.H.; Murry, D.J.; Hohl, R.J. Pharmacodynamic effects of high dose lovastatin in subjects with advanced malignancies. Cancer Chemother. Pharmacol. 2006, 57, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Schafer, W.; Stroh, A.; Berghofer, S.; Seiler, J.; Vey, M.; Kruse, M.L.; Kern, H.F.; Klenk, H.D.; Garten, W. Two independent targeting signals in the cytoplasmic domain determine trans-Golgi network localization and endosomal trafficking of the proprotein convertase furin. EMBO J. 1995, 14, 2424–2435. [Google Scholar] [PubMed]
- Plaimauer, B.; Mohr, G.; Wernhart, W.; Himmelspach, M.; Dorner, F.; Schlokat, U. ‘Shed’ furin: Mapping of the cleavage determinants and identification of its C-terminus. Biochem. J. 2001, 354, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Couture, F.; D’Anjou, F.; Day, R. On the cutting edge of proprotein convertase pharmacology: from molecular concepts to clinical applications. Biomol. Concepts 2011, 2, 421–438. [Google Scholar] [CrossRef] [PubMed]
- De Cicco, R.L.; Bassi, D.E.; Benavides, F.; Conti, C.J.; Klein-Szanto, A.J. Inhibition of proprotein convertases: approaches to block squamous carcinoma development and progression. Mol. Carcinog. 2007, 46, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Sarac, M.S.; Cameron, A.; Lindberg, I. The furin inhibitor hexa-d-arginine blocks the activation of Pseudomonas aeruginosa exotoxin A in vivo. Infect. Immun. 2002, 70, 7136–7139. [Google Scholar] [CrossRef] [PubMed]
- Sarac, M.S.; Peinado, J.R.; Leppla, S.H.; Lindberg, I. Protection against anthrax toxemia by hexa-d-arginine in vitro and in vivo. Infect. Immun. 2004, 72, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Basak, A. Inhibitors of proprotein convertases. J. Mol. Med. (Berl.) 2005, 83, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Shayman, J.A. The design and clinical development of inhibitors of glycosphingolipid synthesis: Will invention be the mother of necessity? Trans. Am. Clin. Climatol. Assoc. 2013, 124, 46–60. [Google Scholar] [PubMed]
- Liu, Y.Y.; Hill, R.A.; Li, Y.T. Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv. Cancer Res. 2013, 117, 59–89. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cellular Action | Targeted Step of Stx Intoxication | Cell Lines Tested | In Vivo Studies | Reference(s) |
|---|---|---|---|---|---|
| CQ | Elevation of pH in acidified organelles | Translocation of A1-moiety to cytosol (predicted) | HEp-2 | - | [98] |
| Baf | V-ATPase inhibitor | Transport to the Golgi | HEp-2 | - | [98] |
| ConA | V-ATPase inhibitor | Transport to the Golgi | HEp-2 | - | [98] |
| Nig | Ionophore that exchanges H+ for monovalent cations | Not determined; later than Golgi | HEp-2 | - | [98] |
| 2DG | Inhibition of glycolysis and protein N-glycosylation; Ca2+ release from the endoplasmic reticulum (ER); Inhibition of Gb3 synthesis | Release of A1-moiety | HEp-2, HT-29, SW480, HeLa | - | [99] |
| FDG | Inhibition of glycolysis and protein N-glycosylation; Ca2+ release from the ER; Inhibition of GlcCer synthesis | Binding; Transport from Golgi to ER; Release of A1-moiety | HEp-2, HT-29, MCF-7, HBMEC | - | [48] |
| Retro-2 substances | Relocalization of Syntaxins 5 and 6 | Transport from endosomes to the Golgi | A459, HeLa | Reduction in mortality rate from 70% to 40% in mice infected with E. coli O104:H4 | [100,101] |
| Mn2+ | Induction of GPP130 oligomerization and its sorting to lysosomes for degradation | Transport from endosomes to the Golgi (no effect on Stx2 transport) | HeLa, Vero | Protection against lethal doses of Stx1 in BALB/c mice; No protection against either Stx1 or Stx2 in CD-1 mice | [65,102,103,104,105] |
| PDMP | Inhibition of GlcCer synthesis | Binding and endocytosis; Transport from endosomes to the Golgi | HEp-2 | - | [43] |
| C-9 | Inhibition of GlcCer synthesis | Not investigated | Human renal tubular epithelial cells, Human glomerular endothelial cells | 50% reduction in mortality rate in rats injected with supernatant from E. coli expressing Stx2 | [106,107,108] |
| HG | Ether lipid precursor | Transport from Golgi to ER | HEp-2, HMEC-1, HBMEC | - | [109] |
| Rosuvastatin | Inhibition of cholesterol biosynthesis and protein prenylation | Transport to the Golgi | ACHN | - | [110] |
| Furin inhibitors | Inhibition of furin | Proteolytic cleavage of the A-moiety | HEp-2 | - | [111,112] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kavaliauskiene, S.; Dyve Lingelem, A.B.; Skotland, T.; Sandvig, K. Protection against Shiga Toxins. Toxins 2017, 9, 44. https://doi.org/10.3390/toxins9020044
Kavaliauskiene S, Dyve Lingelem AB, Skotland T, Sandvig K. Protection against Shiga Toxins. Toxins. 2017; 9(2):44. https://doi.org/10.3390/toxins9020044
Chicago/Turabian StyleKavaliauskiene, Simona, Anne Berit Dyve Lingelem, Tore Skotland, and Kirsten Sandvig. 2017. "Protection against Shiga Toxins" Toxins 9, no. 2: 44. https://doi.org/10.3390/toxins9020044
APA StyleKavaliauskiene, S., Dyve Lingelem, A. B., Skotland, T., & Sandvig, K. (2017). Protection against Shiga Toxins. Toxins, 9(2), 44. https://doi.org/10.3390/toxins9020044