
Asymmetric Cryo-EM Structure of Anthrax Toxin Protective Antigen Pore with Lethal Factor N-Terminal Domain

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cryo-EM Sample Preparation of PA Pore with Three LFN Bound
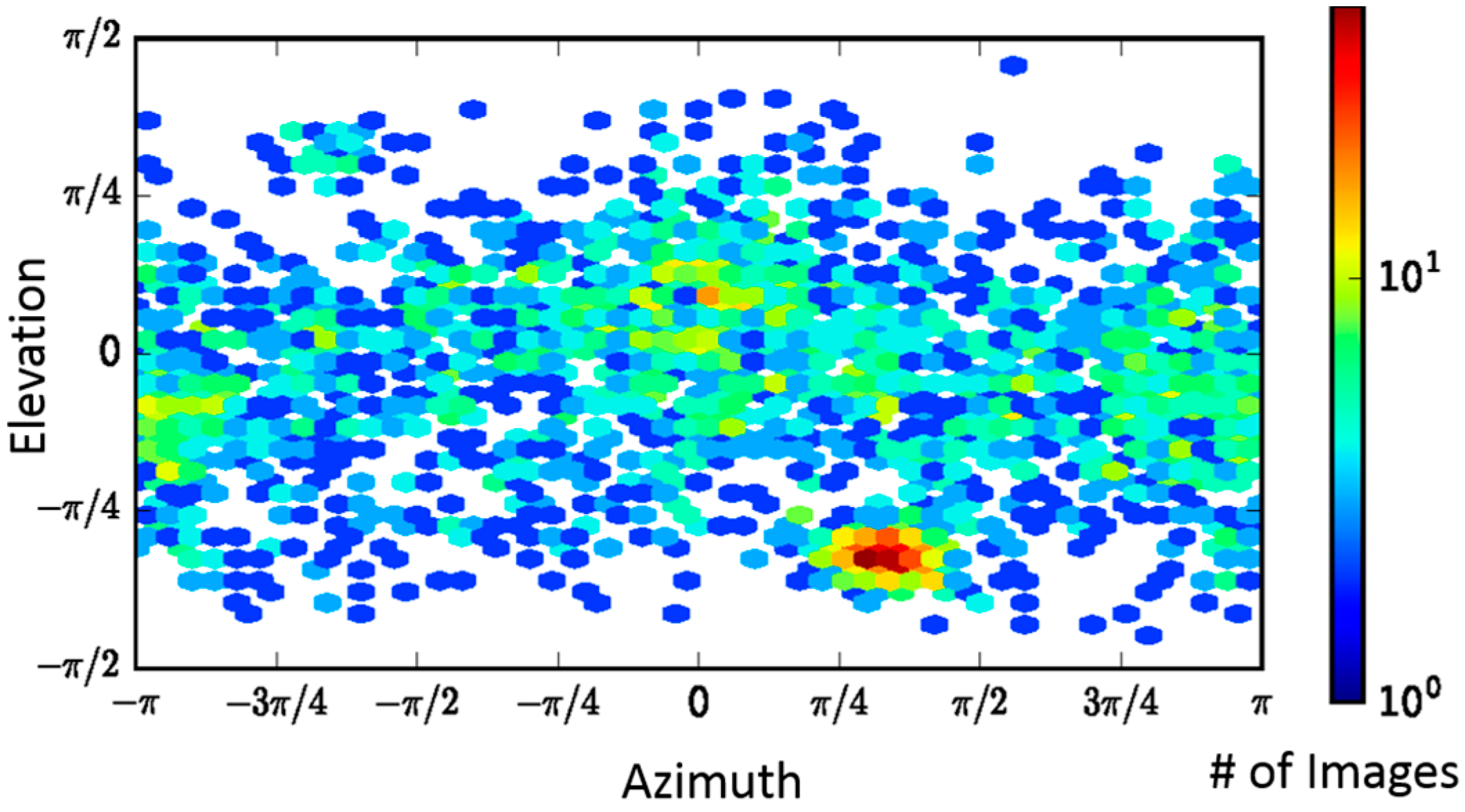
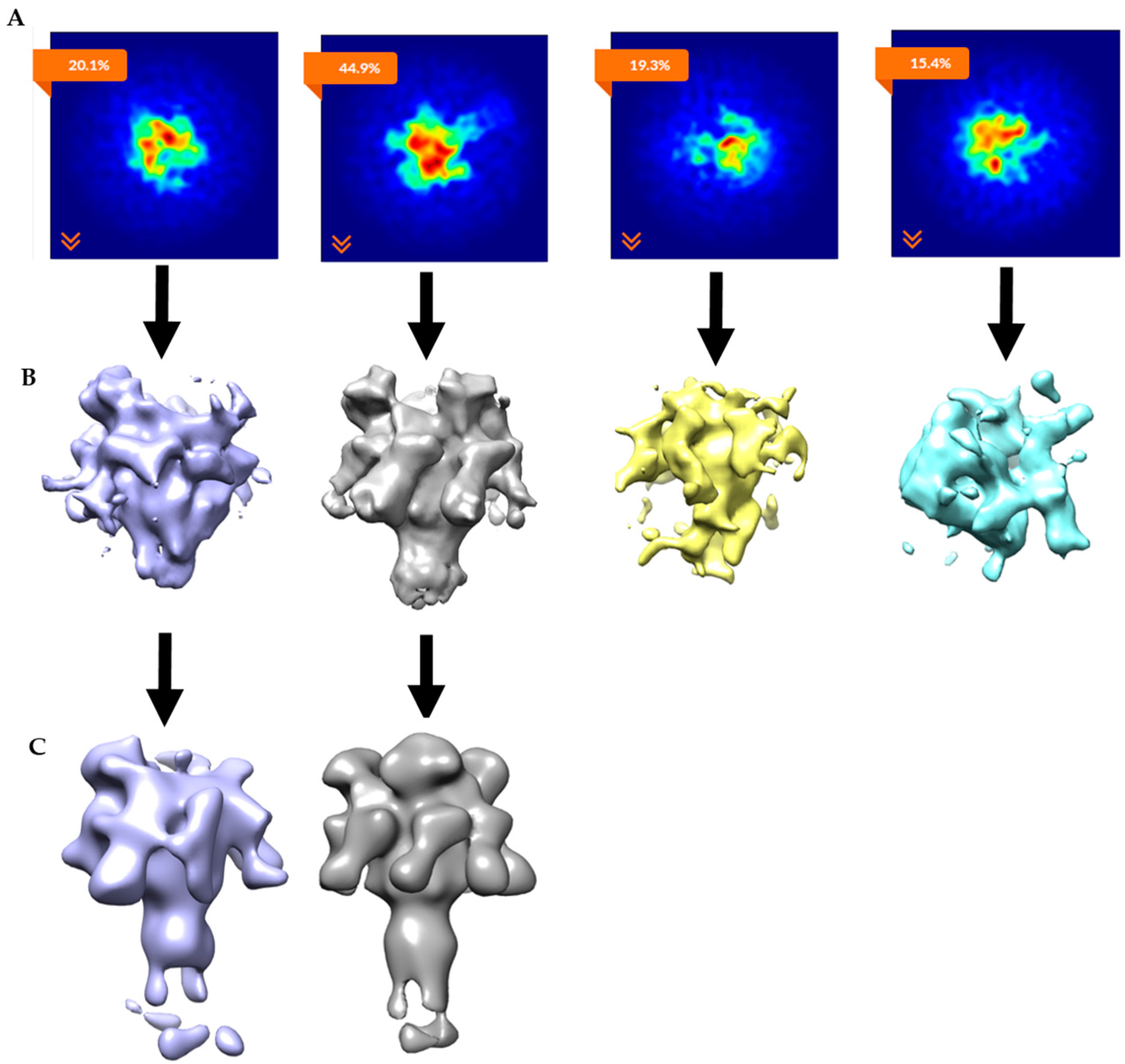
2.2. Single-Particle Analysis of LFN-PA-Nanodisc Complexes
2.3. Constructing Samples with Highly-Populated Singly-Bound LFN-PA for Cryo-EM
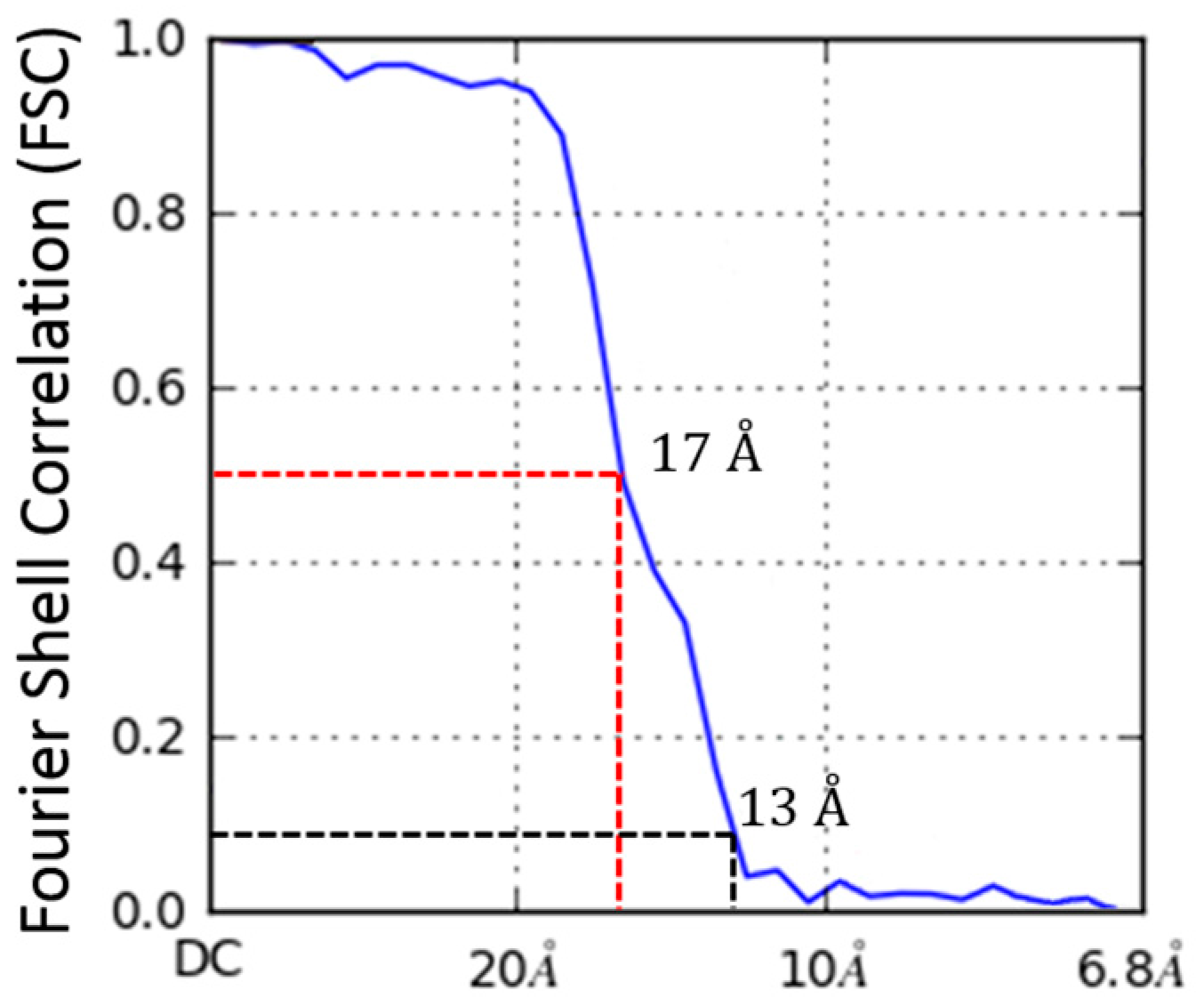
2.4. Molecular Dynamics Flexible Fitting of 3LFN-PA Pore Model into the 17 Å Cryo-EM Density Map
3. Discussion
3.1. Sample Preparation of Highly Pure Complexes
3.2. Initial Cryo-EM Model of 3LFN-PA Pore
4. Conclusions
5. Materials and Methods
5.1. Protein Expression and Purification
5.2. Formation of LFN-PA-Nanodisc Complexes
5.3. Cryo-EM Sample Preparation and Data Collection
5.4. Image Analysis and 3D Reconstruction
5.5. Molecular Dynamics Flexible Fitting of 3LFN-PA
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Lacy, D.B.; Wigelsworth, D.J.; Melnyk, R.A.; Harrison, S.C.; Collier, R.J. Structure of heptameric protective antigen bound to an anthrax toxin receptor: A role for receptor in pH-dependent pore formation. Proc. Natl. Acad. Sci. USA 2004, 101, 13147–13151. [Google Scholar] [CrossRef] [PubMed]
- Santelli, E.; Bankston, L.A.; Leppia, S.H.; Liddington, R.C. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature 2004, 430, 905. [Google Scholar] [CrossRef] [PubMed]
- Wimalasena, D.S.; Janowiak, B.E.; Lovell, S.; Miyagi, M.; Sun, J.; Zhou, H.; Hajduch, J.; Pooput, C.; Kirk, K.L.; Battaile, K.P. Evidence that histidine protonation of receptor-bound anthrax protective antigen is a trigger for pore formation. Biochemistry 2010, 49, 6973–6983. [Google Scholar] [CrossRef] [PubMed]
- Mogridge, J.; Cunningham, K.; Collier, R.J. Stoichiometry of anthrax toxin complexes. Biochemistry 2002, 41, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, I.I.; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A.; Finkelstein, A.; Collier, R.J. Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J. Mol. Biol. 2006, 355, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.; Thoren, K.L.; Krantz, B.A. Role of the α clamp in the protein translocation mechanism of anthrax toxin. J. Mol. Biol. 2015, 427, 3340–3349. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Krantz, B.A. Secondary structure preferences of the anthrax toxin protective antigen translocase. J. Mol. Biol. 2017, 429, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A.; Melnyk, R.A.; Zhang, S.; Juris, S.J.; Lacy, D.B.; Wu, Z.; Finkelstein, A.; Collier, R.J. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science 2005, 309, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, R.A.; Collier, R.J. A loop network within the anthrax toxin pore positions the phenylalanine clamp in an active conformation. Proc. Natl. Acad. Sci. USA 2006, 103, 9802–9807. [Google Scholar] [CrossRef] [PubMed]
- Janowiak, B.E.; Fischer, A.; Collier, R.J. Effects of introducing a single charged residue into the phenylalanine clamp of multimeric anthrax protective antigen. J. Biol. Chem. 2010, 285, 8130–8137. [Google Scholar] [CrossRef] [PubMed]
- Janowiak, B.E.; Jennings-Antipov, L.D.; Collier, R.J. Cys–Cys cross-linking shows contact between the n-terminus of lethal factor and phe427 of the anthrax toxin pore. Biochemistry 2011, 50, 3512–3516. [Google Scholar] [CrossRef] [PubMed]
- Wynia-Smith, S.L.; Brown, M.J.; Chirichella, G.; Kemalyan, G.; Krantz, B.A. Electrostatic ratchet in the protective antigen channel promotes anthrax toxin translocation. J. Biol. Chem. 2012, 287, 43753–43764. [Google Scholar] [CrossRef] [PubMed]
- Pannifer, A.D.; Wong, T.Y.; Schwarzenbacher, R.; Renatus, M.; Petosa, C.; Bienkowska, J.; Lacy, D.B.; Collier, R.J.; Park, S.; Leppla, S.H. Crystal structure of the anthrax lethal factor. Nature 2001, 414, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Feld, G.K.; Thoren, K.L.; Kintzer, A.F.; Sterling, H.J.; Tang, I.I.; Greenberg, S.G.; Williams, E.R.; Krantz, B.A. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat. Struct. Mol. Biol. 2010, 17, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Pentelute, B.L.; Collier, R.J.; Zhou, Z.H. Atomic structure of anthrax pa pore elucidates toxin translocation. Nature 2015, 521, 545. [Google Scholar] [CrossRef] [PubMed]
- Sellman, B.R.; Nassi, S.; Collier, R.J. Point mutations in anthrax protective antigen that block translocation. J. Biol. Chem. 2001, 276, 8371–8376. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lang, A.E.; Aktories, K.; Collier, R.J. Phenylalanine-427 of anthrax protective antigen functions in both pore formation and protein translocation. Proc. Natl. Acad. Sci. USA 2008, 105, 4346–4351. [Google Scholar] [CrossRef] [PubMed]
- Fabre, L.; Santelli, E.; Mountassif, D.; Donoghue, A.; Biswas, A.; Blunck, R.; Hanein, D.; Volkmann, N.; Liddington, R.; Rouiller, I. Structure of anthrax lethal toxin prepore complex suggests a pathway for efficient cell entry. J. Gen. Physiol. 2016, 148, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Pentelute, B.L.; Sharma, O.; Collier, R.J. Chemical dissection of protein translocation through the anthrax toxin pore. Angew. Chem. Int. Ed. Engl. 2011, 50, 2294–2296. [Google Scholar] [CrossRef] [PubMed]
- Gogol, E.; Akkaladevi, N.; Szerszen, L.; Mukherjee, S.; Chollet-Hinton, L.; Katayama, H.; Pentelute, B.; Collier, R.; Fisher, M. Three dimensional structure of the anthrax toxin translocon–lethal factor complex by cryo-electron microscopy. Protein Sci. 2013, 22, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Akkaladevi, N.; Hinton-Chollet, L.; Katayama, H.; Mitchell, J.; Szerszen, L.; Mukherjee, S.; Gogol, E.; Pentelute, B.; Collier, R.; Fisher, M. Assembly of anthrax toxin pore: Lethal-factor complexes into lipid nanodiscs. Protein Sci. 2013, 22, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.; Brock, S.; Akkaladevi, N.; Tally, J.; Mcginn-Straub, W.; Zhang, N.; Gao, P.; Gogol, E.; Pentelute, B.; Collier, R.J. Monitoring the kinetics of the pH-driven transition of the anthrax toxin prepore to the pore by biolayer interferometry and surface plasmon resonance. Biochemistry 2013, 52, 6335–6347. [Google Scholar] [CrossRef] [PubMed]
- Akkaladevi, N.; Mukherjee, S.; Katayama, H.; Janowiak, B.; Patel, D.; Gogol, E.P.; Pentelute, B.L.; Collier, R.J.; Fisher, M.T. Following natures lead: On the construction of membrane-inserted toxins in lipid bilayer nanodiscs. J. Membr. Boil. 2015, 248, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Wang, J.; Tama, F.; Chollet, L.; Gogol, E.; Collier, R.; Fisher, M. Three-dimensional structure of the anthrax toxin pore inserted into lipid nanodiscs and lipid vesicles. Proc. Natl. Acad. Sci. USA 2010, 107, 3453–3457. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Fang, J.; Chittuluru, J.; Asturias, F.J.; Penczek, P.A. Iterative stable alignment and clustering of 2d transmission electron microscope images. Structure 2012, 20, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, M.A.; Punjani, A.; Fleet, D.J. Building Proteins in a Day: Efficient 3D Molecular Reconstruction. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Boston, MA, USA, 7–12 June 2015; pp. 3099–3108. [Google Scholar]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. Cryosparc: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Ripstein, Z.A.; Huang, R.; Augustyniak, R.; Kay, L.E.; Rubinstein, J.L. Structure of a AAA+ unfoldase in the process of unfolding substrate. eLife 2017, 6, e25754. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.S.; Yang, X.; DeCaen, P.G.; Liu, X.; Bulkley, D.; Clapham, D.E.; Cao, E. The structure of the polycystic kidney disease channel pkd2 in lipid nanodiscs. Cell 2016, 167, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, G.; Lopez, F.; Mallardi, A. Effect of detergent concentration on the thermal stability of a membrane protein: The case study of bacterial reaction center solubilized by N,N-dimethyldodecylamine-N-oxide. Biochim. Biophys. Acta 2010, 1804, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Patargias, G.; Bond, P.J.; Deol, S.S.; Sansom, M.S. Molecular dynamics simulations of GlpF in a micelle vs in a bilayer: Conformational dynamics of a membrane protein as a function of environment. J. Phys. Chem. B 2005, 109, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.; Sansom, M.S. One membrane protein, two structures and six environments: A comparative molecular dynamics simulation study of the bacterial outer membrane protein pagp. Mol. Membr. Biol. 2009, 26, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Eddy, M.T.; Su, Y.; Silvers, R.; Andreas, L.; Clark, L.; Wagner, G.; Pintacuda, G.; Emsley, L.; Griffin, R.G. Lipid bilayer-bound conformation of an integral membrane beta barrel protein by multidimensional MAS NMR. J. Biomol. NMR 2015, 61, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Elad, N.; Clare, D.K.; Saibil, H.R.; Orlova, E.V. Detection and separation of heterogeneity in molecular complexes by statistical analysis of their two-dimensional projections. J. Struct. Biol. 2008, 162, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.; Jiang, M.; Roth, A.; Puchalla, J.; Zhang, J.; Rye, H.S. Groel actively stimulates folding of the endogenous substrate protein pepq. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H.R.; Ranson, N.A. The chaperonin folding machine. Trends Biochem. Sci. 2002, 27, 627–632. [Google Scholar] [CrossRef]
- Huang, R.; Ripstein, Z.A.; Augustyniak, R.; Lazniewski, M.; Ginalski, K.; Kay, L.E.; Rubinstein, J.L. Unfolding the mechanism of the aaa+ unfoldase vat by a combined cryo-em, solution nmr study. Proc. Natl. Acad. Sci. USA 2016, 113, 4190–4199. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.J.; Elliott, J.L.; Collier, R.J. Anthrax protective antigen: Prepore-to-pore conversion. Biochemistry 1999, 38, 10432–10441. [Google Scholar] [CrossRef] [PubMed]
- Wigelsworth, D.J.; Krantz, B.A.; Christensen, K.A.; Lacy, D.B.; Juris, S.J.; Collier, R.J. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, cmg2, with protective antigen. J. Biol. Chem. 2004, 279, 23349–23356. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.; Grinkova, Y.; Bayburt, T.; Denisov, I.; Zolnerciks, J.; Atkins, W.; Sligar, S. Chapter eleven-reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 2009, 464, 211–231. [Google Scholar] [PubMed]
- Denisov, I.; Grinkova, Y.; Lazarides, A.; Sligar, S. Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 2004, 126, 3477–3487. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Peng, L.; Baldwin, P.R.; Mann, D.S.; Jiang, W.; Rees, I.; Ludtke, S.J. Eman2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 2007, 157, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Lee, J.; Singharoy, A.; McGreevy, R.; Schulten, K.; Im, W. Charmm-gui mdff/xmdff utilizer for molecular dynamics flexible fitting simulations in various environments. J. Phys. Chem. B 2016, 121, 3718–3723. [Google Scholar] [CrossRef] [PubMed]
- Birmanns, S.; Rusu, M.; Wriggers, W. Using sculptor and situs for simultaneous assembly of atomic components into low-resolution shapes. J. Struct. Biol. 2011, 173, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Wahle, M.; Wriggers, W. Multi-scale visualization of molecular architecture using real-time ambient occlusion in sculptor. PLoS Comput. Biol. 2015, 11, e1004516. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Trabuco, L.G.; Villa, E.; Mitra, K.; Frank, J.; Schulten, K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 2008, 16, 673–683. [Google Scholar] [CrossRef] [PubMed]











© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machen, A.J.; Akkaladevi, N.; Trecazzi, C.; O’Neil, P.T.; Mukherjee, S.; Qi, Y.; Dillard, R.; Im, W.; Gogol, E.P.; White, T.A.; et al. Asymmetric Cryo-EM Structure of Anthrax Toxin Protective Antigen Pore with Lethal Factor N-Terminal Domain. Toxins 2017, 9, 298. https://doi.org/10.3390/toxins9100298
Machen AJ, Akkaladevi N, Trecazzi C, O’Neil PT, Mukherjee S, Qi Y, Dillard R, Im W, Gogol EP, White TA, et al. Asymmetric Cryo-EM Structure of Anthrax Toxin Protective Antigen Pore with Lethal Factor N-Terminal Domain. Toxins. 2017; 9(10):298. https://doi.org/10.3390/toxins9100298
Chicago/Turabian StyleMachen, Alexandra J., Narahari Akkaladevi, Caleb Trecazzi, Pierce T. O’Neil, Srayanta Mukherjee, Yifei Qi, Rebecca Dillard, Wonpil Im, Edward P. Gogol, Tommi A. White, and et al. 2017. "Asymmetric Cryo-EM Structure of Anthrax Toxin Protective Antigen Pore with Lethal Factor N-Terminal Domain" Toxins 9, no. 10: 298. https://doi.org/10.3390/toxins9100298
APA StyleMachen, A. J., Akkaladevi, N., Trecazzi, C., O’Neil, P. T., Mukherjee, S., Qi, Y., Dillard, R., Im, W., Gogol, E. P., White, T. A., & Fisher, M. T. (2017). Asymmetric Cryo-EM Structure of Anthrax Toxin Protective Antigen Pore with Lethal Factor N-Terminal Domain. Toxins, 9(10), 298. https://doi.org/10.3390/toxins9100298