
Cellular Entry of the Diphtheria Toxin Does Not Require the Formation of the Open-Channel State by Its Translocation Domain


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
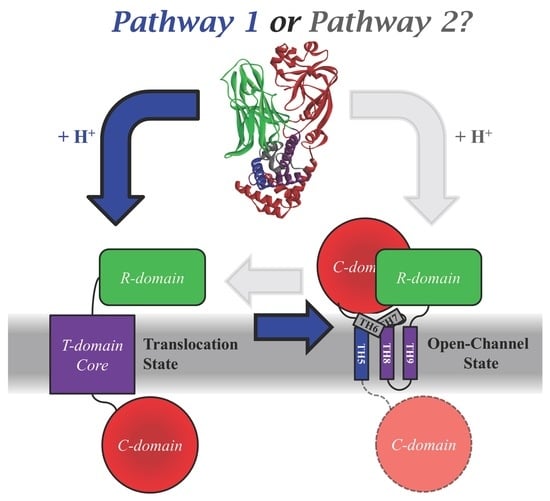
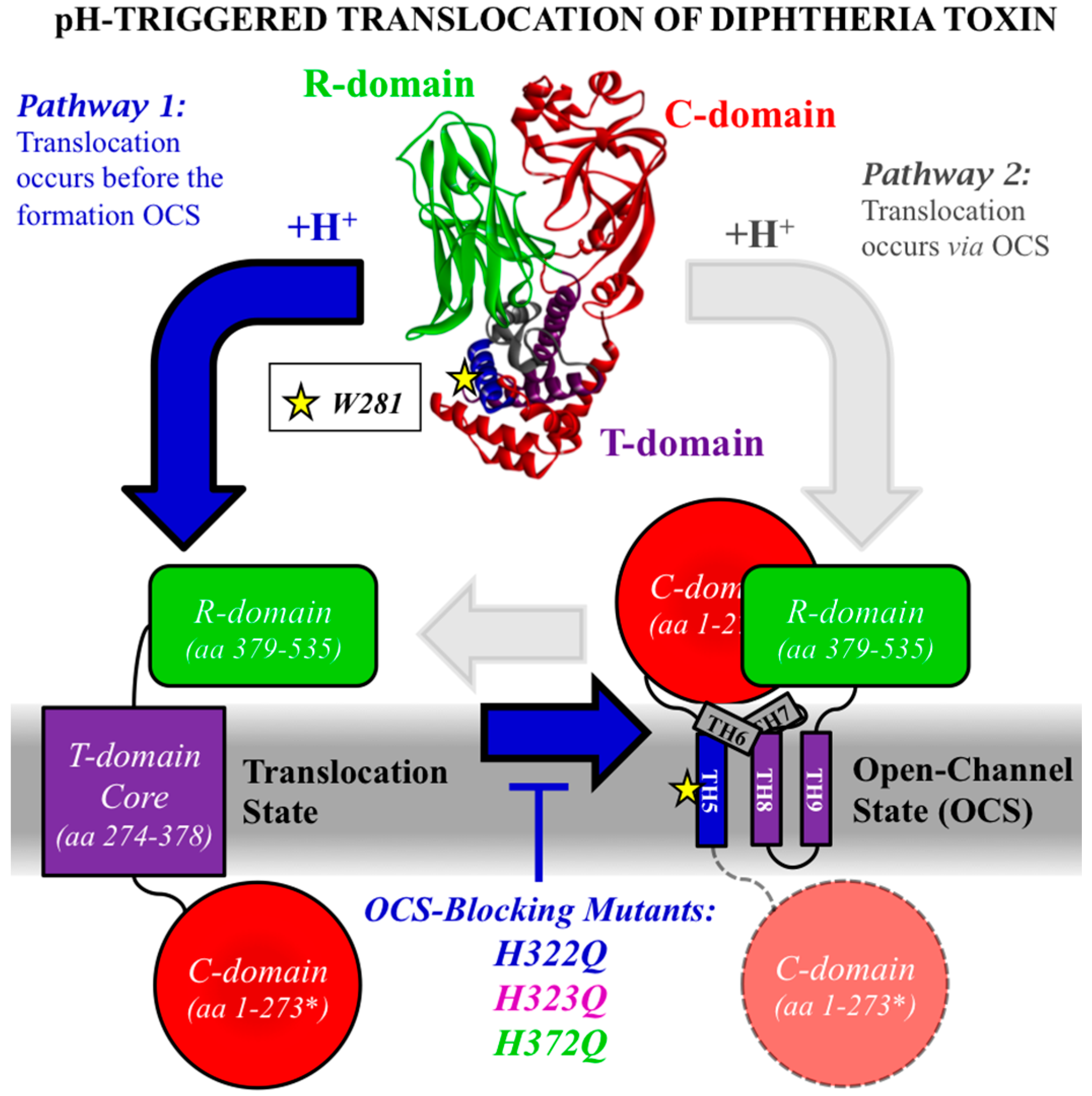
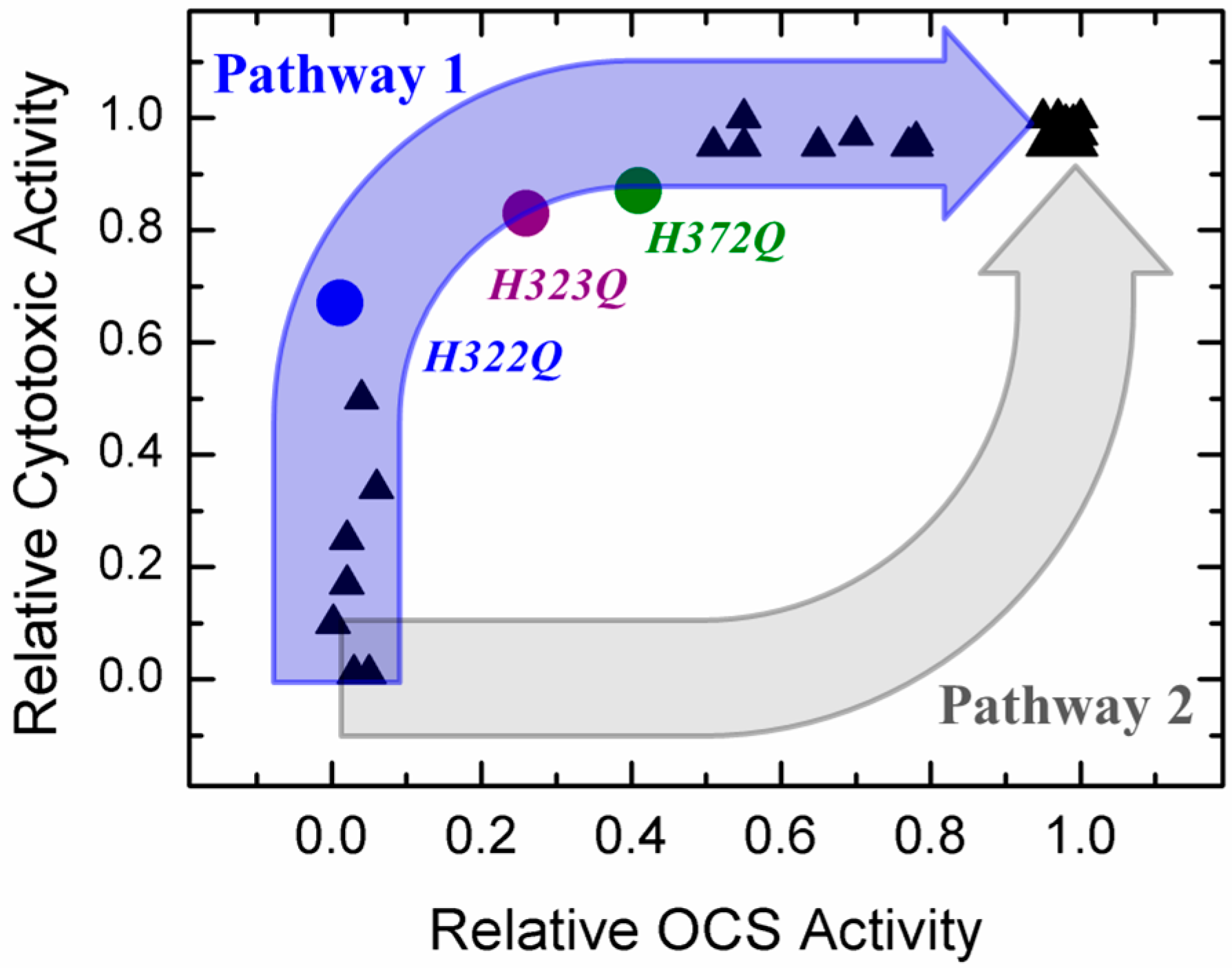
2.1. Comparing the Two Translocation Pathways
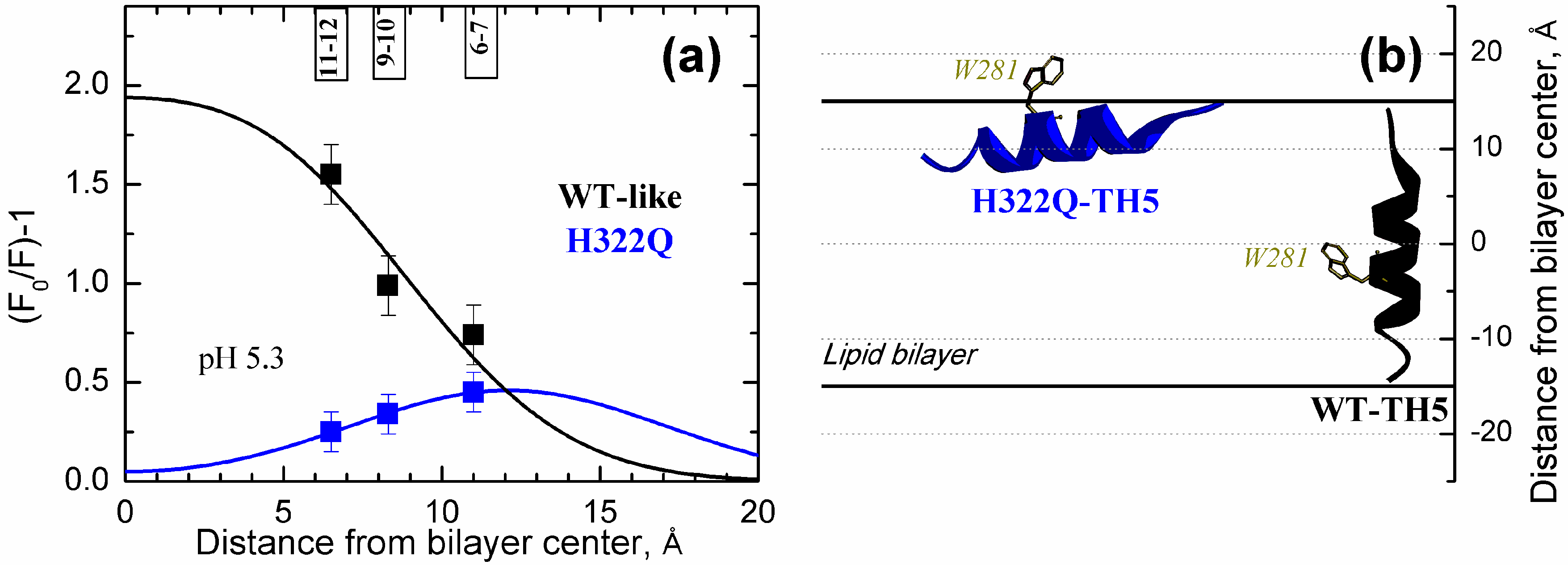
2.2. Spectroscopic Evidence for the Difference in TH5 Topology in WT and in OCS-Blocking Mutant H322Q
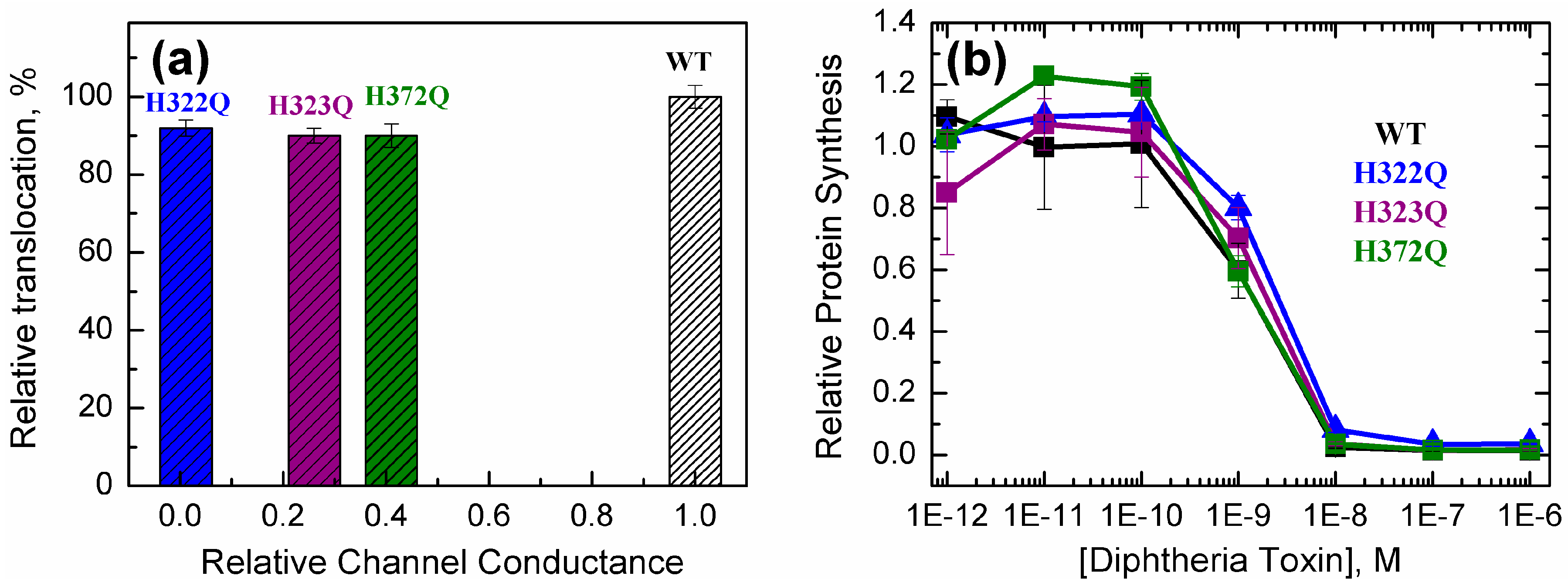
2.3. Translocation Activity of OCS-Blocking Mutants of the T-Domain
2.4. OCS: Critical Intermediate or Byproduct of Translocation
3. Conclusions and Perspectives
- Is it possible that these mutants take advantage, for some reason, of an entry pathway alternative to that of the WT toxin? While such an option is possible, it seems rather unlikely, because these mutants also appear active in a simplified in vitro translocation assay performed in a reductionist system of artificial lipid vesicles without a transbilayer electrical potential (Figure 4a);
- Is it possible that the number of molecules in the OCS conformation is only a minor fraction of the entire population in model experiments? Our spectroscopic data indicate otherwise, suggesting a clear correlation between the ability of helix TH5 to insert in the OCS conformation or its precursor even in the absence of transbilayer potential (Figure 2 and Figure 3 and [38]);
- What is the mechanism of the translocation? Clearly more model and cellular studies will be necessary to fully answer this question. One possibility may involve the formation of a transient passageway due to the perturbation caused by the T-domain refolding on bilayer interface. Deciphering the molecular mechanism of this enigmatic system is especially important in light of the potential use for diphtheria toxin T-domain as a molecular vehicle for targeted drug delivery.
4. Materials and Methods
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murphy, J.R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011, 3, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.C.; Aktories, K.; Barbieri, J.T. Novel bacterial ADP-ribosylating toxins: Structure and function. Nat. Rev. Microbiol. 2014, 12, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schulte, W.; Pink, D.; Phipps, K.; Zijlstra, A.; Lewis, J.D.; Waisman, D.M. Sensitivity of cancer cells to truncated diphtheria toxin. PLoS ONE 2010, 5, e10498. [Google Scholar] [CrossRef] [PubMed]
- Wild, R.; Yokoyama, Y.; Dings, R.P.; Ramakrishnan, S. VEGF-DT385 toxin conjugate inhibits mammary adenocarcinoma development in a transgenic mouse model of spontaneous tumorigenesis. Breast Cancer Res. Treat. 2004, 85, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Urieto, J.O.; Liu, T.; Black, J.H.; Cohen, K.A.; Hall, P.D.; Willingham, M.C.; Pennell, L.K.; Hogge, D.E.; Kreitman, R.J.; Frankel, A.E. Expression and purification of the recombinant diphtheria fusion toxin DT388IL3 for phase i clinical trials. Protein Expr. Purif. 2004, 33, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Turturro, F. Denileukin diftitox: A biotherapeutic paradigm shift in the treatment of lymphoid-derived disorders. Expert Rev. Anticancer Ther. 2007, 7, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Olson, T.A.; Bautch, V.L.; Mohanraj, D. Vascular endothelial growth factor-toxin conjugate specifically inhibits KDR/flk-1-positive endothelial cell proliferation in vitro and angiogenesis in vivo. Cancer Res. 1996, 56, 1324–1330. [Google Scholar] [PubMed]
- Ramage, J.G.; Vallera, D.A.; Black, J.H.; Aplan, P.D.; Kees, U.R.; Frankel, A.E. The diphtheria toxin/urokinase fusion protein (DTAT) is selectively toxic to CD87 expressing leukemic cells. Leuk. Res. 2003, 27, 79–84. [Google Scholar] [CrossRef]
- Murphy, J.R.; Bishai, W.; Borowski, M.; Miyanohara, A.; Boyd, J.; Nagle, S. Genetic construction, expression, and melanoma-selective cytotoxicity of a diphtheria toxin-related alpha-melanocyte-stimulating hormone fusion protein. Proc. Natl. Acad. Sci. USA 1986, 83, 8258–8262. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J. Immunotoxins for targeted cancer therapy. AAPS J. 2006, 8, E532–E551. [Google Scholar] [CrossRef] [PubMed]
- Hogge, D.E.; Yalcintepe, L.; Wong, S.H.; Gerhard, B.; Frankel, A.E. Variant diphtheria toxin-interleukin-3 fusion proteins with increased receptor affinity have enhanced cytotoxicity against acute myeloid leukemia progenitors. Clin. Cancer Res. 2006, 12, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.D.; Willingham, M.C.; Kreitman, R.J.; Frankel, A.E. DT388-GM-CSF, a novel fusion toxin consisting of a truncated diphtheria toxin fused to human granulocyte-macrophage colony-stimulating factor, prolongs host survival in a scid mouse model of acute myeloid leukemia. Leukemia 1999, 13, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Feuring-Buske, M.; Frankel, A.; Gerhard, B.; Hogge, D. Variable cytotoxicity of diphtheria toxin 388-granulocyte-macrophage colony-stimulating factor fusion protein for acute myelogenous leukemia stem cells. Exp. Hematol. 2000, 28, 1390–1400. [Google Scholar] [CrossRef]
- Duvic, M.; Talpur, R. Optimizing denileukin diftitox (ontak) therapy. Future Oncol. 2008, 4, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.A.; Liu, T.F.; Cline, J.M.; Wagner, J.D.; Hall, P.D.; Frankel, A.E. Toxicology and pharmacokinetics of DT388IL3, a fusion toxin consisting of a truncated diphtheria toxin (DT388) linked to human interleukin 3 (IL3), in cynomolgus monkeys. Leuk. Lymphoma 2004, 45, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Black, J.H.; McCubrey, J.A.; Willingham, M.C.; Ramage, J.; Hogge, D.E.; Frankel, A.E. Diphtheria toxin-interleukin-3 fusion protein (DT(388)IL3) prolongs disease-free survival of leukemic immunocompromised mice. Leukemia 2003, 17, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, Y.K.; Andreev, O.A.; Lehnert, U.; Engelman, D.M. Translocation of molecules into cells by pH-dependent insertion of a transmembrane helix. Proc. Natl. Acad. Sci. USA 2006, 103, 6460–6465. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, Y.K.; Segala, M.; Andreev, O.A.; Engelman, D.M. A monomeric membrane peptide that lives in three worlds: In solution, attached to, and inserted across lipid bilayers. Biophys. J. 2007, 93, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Andreev, O.A.; Engelman, D.M.; Reshetnyak, Y.K. Targeting acidic diseased tissue: New technology based on use of the pH (Low) Insertion Peptide (pHLIP). Chim. Oggi 2009, 27, 34–37. [Google Scholar] [PubMed]
- Segala, J.; Engelman, D.M.; Reshetnyak, Y.K.; Andreev, O.A. Accurate analysis of tumor margins using a fluorescent pH Low Insertion Peptide (pHLIP). Int. J. Mol. Sci. 2009, 10, 3478–3487. [Google Scholar] [CrossRef] [PubMed]
- Vavere, A.L.; Biddlecombe, G.B.; Spees, W.M.; Garbow, J.R.; Wijesinghe, D.; Andreev, O.A.; Engelman, D.M.; Reshetnyak, Y.K.; Lewis, J.S. A novel technology for the imaging of acidic prostate tumors by positron emission tomography. Cancer Res. 2009, 69, 4510–4516. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, Y.K.; Yao, L.; Zheng, S.; Kuznetsov, S.; Engelman, D.M.; Andreev, O.A. Measuring tumor aggressiveness and targeting metastatic lesions with fluorescent pHLIP. Mol. Imaging Biol. 2011, 13, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Andreev, O.A.; Engelman, D.M.; Reshetnyak, Y.K. pH-sensitive membrane peptides (pHLIPs) as a novel class of delivery agents. Mol. Membr. Biol. 2010, 27, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Sosunov, E.A.; Anyukhovsky, E.P.; Sosunov, A.A.; Moshnikova, A.; Wijesinghe, D.; Engelman, D.M.; Reshetnyak, Y.K.; Andreev, O.A. pH (Low) Insertion Peptide (pHLIP) targets ischemic myocardium. Proc. Natl. Acad. Sci. USA 2013, 110, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Weerakkody, D.; Moshnikova, A.; Thakur, M.S.; Moshnikova, V.; Daniels, J.; Engelman, D.M.; Andreev, O.A.; Reshetnyak, Y.K. Family of pH (Low) Insertion Peptides for tumor targeting. Proc. Natl. Acad. Sci. USA 2013, 110, 5834–5839. [Google Scholar] [CrossRef] [PubMed]
- Andreev, O.A.; Engelman, D.M.; Reshetnyak, Y.K. Targeting diseased tissues by pHLIP insertion at low cell surface pH. Front. Physiol. 2014, 5, 97. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Kachel, K.; Kim, H.; Malenbaum, S.E.; Collier, R.J.; London, E. Interaction of diphtheria toxin T domain with molten globule-like proteins and its implications for translocation. Science 1999, 284, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Ladokhin, A.S. pH-triggered conformational switching along the membrane insertion pathway of the diphtheria toxin T-domain. Toxins 2013, 5, 1362–1380. [Google Scholar] [CrossRef] [PubMed]
- Ladokhin, A.S.; Legmann, R.; Collier, R.J.; White, S.H. Reversible refolding of the diphtheria toxin T-domain on lipid membranes. Biochemistry 2004, 43, 7451–7458. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, A.; Posokhov, Y.O.; Rodnin, M.V.; Ladokhin, A.S. Kinetic intermediate reveals staggered pH-dependent transitions along the membrane insertion pathway of the diphtheria toxin T-domain. Biochemistry 2009, 48, 7584–7594. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Uribe, M.; Rodnin, M.V.; Ojemalm, K.; Holgado, A.; Kyrychenko, A.; Nilsson, I.; Posokhov, Y.O.; Makhatadze, G.; von Heijne, G.; Ladokhin, A.S. Thermodynamics of membrane insertion and refolding of the diphtheria toxin T-domain. J. Membr. Biol. 2015, 248, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Rodnin, M.V.; Kyrychenko, A.; Kienker, P.; Sharma, O.; Posokhov, Y.O.; Collier, R.J.; Finkelstein, A.; Ladokhin, A.S. Conformational switching of the diphtheria toxin T domain. J. Mol. Biol. 2010, 402, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kurnikov, I.V.; Kyrychenko, A.; Flores-Canales, J.C.; Rodnin, M.V.; Simakov, N.; Vargas-Uribe, M.; Posokhov, Y.O.; Kurnikova, M.; Ladokhin, A.S. Ph-triggered conformational switching of the diphtheria toxin T-domain: The roles of N-terminal histidines. J. Mol. Biol. 2013, 425, 2752–2764. [Google Scholar] [CrossRef] [PubMed]
- Rodnin, M.V.; Li, J.; Gross, M.L.; Ladokhin, A.S. The pH-dependent trigger in diphtheria toxin T domain comes with a safety latch. Biophys. J. 2016, 111, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Perier, A.; Chassaing, A.; Raffestin, S.; Pichard, S.; Masella, M.; Menez, A.; Forge, V.; Chenal, A.; Gillet, D. Concerted protonation of key histidines triggers membrane interaction of the diphtheria toxin T domain. J. Biol. Chem. 2007, 282, 24239–24245. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, C.; Rodnin, M.V.; Vargas-Uribe, M.; McCluskey, A.J.; Flores-Canales, J.C.; Kurnikova, M.; Ladokhin, A.S. Role of acidic residues in helices TH8–TH9 in membrane interactions of the diphtheria toxin T domain. Toxins 2015, 7, 1303–1323. [Google Scholar] [CrossRef] [PubMed]
- Rodnin, M.V.; Kyrychenko, A.; Kienker, P.; Sharma, O.; Vargas-Uribe, M.; Collier, R.J.; Finkelstein, A.; Ladokhin, A.S. Replacement of C-terminal histidines uncouples membrane insertion and translocation in diphtheria toxin T-domain. Biophys. J. 2011, 101, L41–L43. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Uribe, M.; Rodnin, M.V.; Kienker, P.; Finkelstein, A.; Ladokhin, A.S. Crucial role of H322 in folding of the diphtheria toxin T-domain into the Open-Channel State. Biochemistry 2013, 52, 3457–3463. [Google Scholar] [CrossRef] [PubMed]
- Kienker, P.K.; Wu, Z.; Finkelstein, A. Mapping the membrane topography of the TH6–TH7 segment of the diphtheria toxin T-domain channel. J. Gen. Physiol. 2015, 145, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Kienker, P.K.; Wu, Z.; Finkelstein, A. Topography of the TH5 segment in the diphtheria toxin T-domain channel. J. Membr. Biol. 2016, 249, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Donovan, J.J.; Simon, M.I.; Draper, R.K.; Montal, M. Diphtheria toxin forms transmembrane channels in planar lipid bilayers. Proc. Natl. Acad. Sci. USA 1981, 78, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.J.; Choe, S.; Eisenberg, D. Refined structure of dimeric diphtheria toxin at 2.0 Å resolution. Protein Sci. 1994, 3, 1444–1463. [Google Scholar] [CrossRef] [PubMed]
- Senzel, L.; Gordon, M.; Blaustein, R.O.; Oh, K.J.; Collier, R.J.; Finkelstein, A. Topography of diphtheria toxin’s T domain in the open channel state. J. Gen. Physiol. 2000, 115, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Zhan, H.; Cui, C.; Hideg, K.; Collier, R.J.; Hubbell, W.L. Organization of diphtheria toxin T domain in bilayers: A site-directed spin labeling study. Science 1996, 273, 810–812. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A.; Silverman, J.A.; Collier, R.J.; Finkelstein, A. Structure-function relationships in diphtheria toxin channels: III. Residues which affect the cis pH dependence of channel conductance. J. Membr. Biol. 1994, 137, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.A.; Mindell, J.A.; Finkelstein, A.; Shen, W.H.; Collier, R.J. Mutational analysis of the helical hairpin region of diphtheria toxin transmembrane domain. J. Biol. Chem. 1994, 269, 22524–22532. [Google Scholar] [PubMed]
- Silverman, J.A.; Mindell, J.A.; Zhan, H.; Finkelstein, A.; Collier, R.J. Structure-function relationships in diphtheria toxin channels: I. Determining a minimal channel-forming domain. J. Membr. Biol. 1994, 137, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Kaul, P.; Silverman, J.; Shen, W.H.; Blanke, S.R.; Huynh, P.D.; Finkelstein, A.; Collier, R.J. Roles of Glu 349 and Asp 352 in membrane insertion and translocation by diphtheria toxin. Protein Sci. 1996, 5, 687–892. [Google Scholar] [CrossRef] [PubMed]
- Rodnin, M.V.; Ladokhin, A.S. Membrane translocation assay based on proteolytic cleavage: Application to diphtheria toxin T domain. Biochim. Biophys. Acta 2015, 1848, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Falnes, P.O.; Madshus, I.H.; Sandvig, K.; Olsnes, S. Replacement of negative by positive charges in the presumed membrane-inserted part of diphtheria toxin B fragment. Effect on membrane translocation and on formation of cation channels. J. Biol. Chem. 1992, 267, 12284–12290. [Google Scholar] [PubMed]
- London, E.; Ladokhin, A.S. Measuring the depth of amino acid residues in membrane-inserted peptides by fluorescence quenching. Curr. Top. Membr. 2002, 52, 89–115. [Google Scholar]
- Ladokhin, A.S. Measuring membrane penetration with depth-dependent fluorescence quenching: Distribution analysis is coming of age. Biochim. Biophys. Acta 2014, 1838, 2289–2295. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, A.; Posokhov, Y.O.; Vargas-Uribe, M.; Ghatak, C.; Rodnin, M.V.; Ladokhin, A.S. Fluorescence applications for structural and thermodynamic studies of membrane protein insertion. In Reviews in Fluorescence 2016; Geddes, C.D., Ed.; Springer: Cham, Switzerland, 2017; pp. 243–274. [Google Scholar]
- McIntosh, T.J.; Holloway, P.W. Determination of the depth of bromine atoms in bilayers formed from bromolipid probes. Biochemistry 1987, 26, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Ladokhin, A.S. Analysis of protein and peptide penetration into membranes by depth-dependent fluorescence quenching: Theoretical considerations. Biophys. J. 1999, 76, 946–955. [Google Scholar] [CrossRef]
- Ladokhin, A.S. Evaluation of lipid exposure of tryptophan residues in membrane peptides and proteins. Anal. Biochem. 1999, 276, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Gnanasambandam, R.; Ghatak, C.; Yasmann, A.; Nishizawa, K.; Sachs, F.; Ladokhin, A.S.; Sukharev, S.I.; Suchyna, T.M. GsMTx4: Mechanism of inhibiting mechanosensitive ion channels. Biophys. J. 2017, 112, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Blanke, S.R.; Milne, J.C.; Benson, E.L.; Collier, R.J. Fused polycationic peptide mediates delivery of diphtheria toxin A chain to the cytosol in the presence of anthrax protective antigen. Proc. Natl. Acad. Sci. USA 1996, 93, 8437–8442. [Google Scholar] [CrossRef] [PubMed]
- Hope, M.J.; Bally, M.B.; Mayer, L.D.; Janoff, A.S.; Cullis, P.R. Generation of multilamellar and unilamellar phospholipid vesicles. Chem. Phys. Lipids 1986, 40, 89–107. [Google Scholar] [CrossRef]
- Mayer, L.D.; Hope, M.J.; Cullis, P.R. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta 1986, 858, 161–168. [Google Scholar] [CrossRef]
- Barbieri, J.T.; Collier, R.J. Expression of a mutant, full-length form of diphtheria toxin in Escherichia coli. Infect Immun. 1987, 55, 1647–1651. [Google Scholar] [PubMed]
- Zhan, H.; Elliott, J.L.; Shen, W.H.; Huynh, P.D.; Finkelstein, A.; Collier, R.J. Effects of mutations in proline 345 on insertion of diptheria toxin into model membranes. J. Membr. Biol. 1999, 167, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Ladokhin, A.S.; Jayasinghe, S.; White, S.H. How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal. Biochem. 2000, 285, 235–245. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ladokhin, A.S.; Vargas-Uribe, M.; Rodnin, M.V.; Ghatak, C.; Sharma, O. Cellular Entry of the Diphtheria Toxin Does Not Require the Formation of the Open-Channel State by Its Translocation Domain. Toxins 2017, 9, 299. https://doi.org/10.3390/toxins9100299
Ladokhin AS, Vargas-Uribe M, Rodnin MV, Ghatak C, Sharma O. Cellular Entry of the Diphtheria Toxin Does Not Require the Formation of the Open-Channel State by Its Translocation Domain. Toxins. 2017; 9(10):299. https://doi.org/10.3390/toxins9100299
Chicago/Turabian StyleLadokhin, Alexey S., Mauricio Vargas-Uribe, Mykola V. Rodnin, Chiranjib Ghatak, and Onkar Sharma. 2017. "Cellular Entry of the Diphtheria Toxin Does Not Require the Formation of the Open-Channel State by Its Translocation Domain" Toxins 9, no. 10: 299. https://doi.org/10.3390/toxins9100299
APA StyleLadokhin, A. S., Vargas-Uribe, M., Rodnin, M. V., Ghatak, C., & Sharma, O. (2017). Cellular Entry of the Diphtheria Toxin Does Not Require the Formation of the Open-Channel State by Its Translocation Domain. Toxins, 9(10), 299. https://doi.org/10.3390/toxins9100299