
G-Protein Coupled Receptors Targeted by Analgesic Venom Peptides


Abstract
:1. Introduction
2. GPCR Modulation of Nociception
2.1. GPCR Signalling Pathways
2.2. Role of GPCRs in Acute Pain
2.2.1. Peripheral Nociception
2.2.2. Central Processing
2.3. Role of GPCRs in Pathological Pain
2.4. GPCRs as Analgesic Targets
3. Analgesic Venom Peptides Targeting GPCRs
3.1. Development of Venom Peptides as Analgesic Drugs
3.2. Conopeptides Targeting GPCRs with In-Vivo Analgesic Efficacy
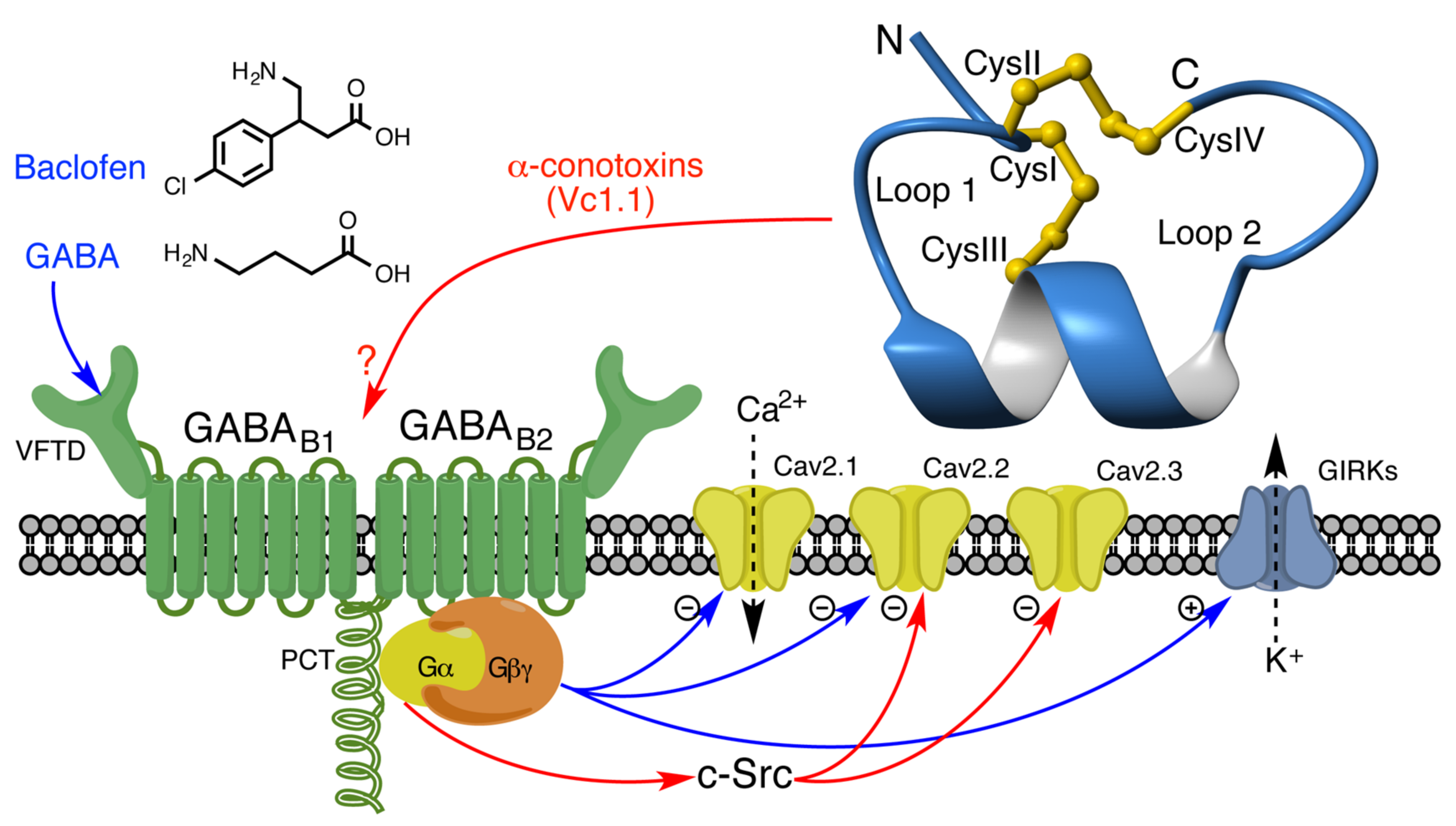
3.2.1. GABAB Receptor Targeted by α-Conotoxins
3.2.2. Neurotensin Receptors Targeted by Contulakin-G
3.2.3. κ-Opioid Receptor Targeted by Conorphins
3.3. Other GPCRs Targeted by Venom Peptides with Potential Analgesic Properties
4. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.R. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature 1996, 380, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Tedford, H.W.; Zamponi, G.W. Direct G protein modulation of Cav2 calcium channels. Pharmacol. Rev. 2006, 58, 837–862. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Ma, Q. Nociceptors—Noxious stimulus detectors. Neuron 2007, 55, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.S.; Gebhart, G.F. Nociceptor sensitization in pain pathogenesis. Nat. Med. 2010, 16, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.-L.; Wu, Z.-Z.; Zhou, H.-Y.; Chen, S.-R.; Zhang, H.-M.; Li, D.-P. Modulation of Pain Transmission by G Protein-Coupled Receptors. Pharmacol. Ther. 2008, 117, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Stone, L.S.; Molliver, D.C. In Search of Analgesia: Emerging Poles of GPCRs in Pain. Mol. Interv. 2009, 9, 234–251. [Google Scholar] [CrossRef] [PubMed]
- Geppetti, P.; Veldhuis, N.A.; Lieu, T.; Bunnett, N.W. G Protein-Coupled Receptors: Dynamic Machines for Signaling Pain and Itch. Neuron 2015, 88, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Kuner, R. Central mechanisms of pathological pain. Nat. Med. 2010, 16, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Altin, M.; Duenas, H.; Alev, L. The Role of Descending Inhibitory Pathways on Chronic Pain Modulation and Clinical Implications. Pain Pract. 2014, 14, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Binder, A.; Wasner, G. Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010, 9, 807–819. [Google Scholar] [CrossRef]
- Van Hecke, O.; Torrance, N.; Smith, B.H. Chronic pain epidemiology and its clinical relevance. Br. J. Anaesth. 2013, 111, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, R.H.; O’Connor, A.B.; Backonja, M.; Farrar, J.T.; Finnerup, N.B.; Jensen, T.S.; Kalso, E.A.; Loeser, J.D.; Miaskowski, C.; Nurmikko, T.J.; et al. Pharmacologic management of neuropathic pain: Evidence-based recommendations. Pain 2007, 132, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.H.; Torrance, N. Epidemiology of neuropathic pain. Pain Manag. 2011, 1, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.H.; Torrance, N. Epidemiology of Neuropathic Pain and Its Impact on Quality of Life. Curr. Pain Headche Rep. 2012, 16, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Gaskin, D.J.; Richard, P. The economic costs of pain in the United States. J. Pain 2012, 13, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Cesare, P.; McNaughton, P. A Novel Heat-Activated Current in Nociceptive Neurons and Its Sensitization by Bradykinin. Proc. Natl. Acad. Sci. USA 1996, 93, 15435–15439. [Google Scholar] [CrossRef] [PubMed]
- Pethő, G.; Reeh, P.W. Sensory and Signaling Mechanisms of Bradykinin, Eicosanoids, Platelet-Activating Factor, and Nitric Oxide in Peripheral Nociceptors. Physiol. Rev. 2012, 92, 1699–1775. [Google Scholar] [CrossRef] [PubMed]
- Aley, K.O.; McCarter, G.; Levine, J.D. Nitric Oxide Signaling in Pain and Nociceptor Sensitization in the Rat. J. Neurosci. 1998, 18, 7008–7014. [Google Scholar] [PubMed]
- Rosa, A.C.; Fantozzi, R. The role of histamine in neurogenic inflammation. Br. J. Pharmacol. 2013, 170, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Wada, M.; Masu, M. Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc. Natl. Acad. Sci. USA 2001, 98, 6951–6956. [Google Scholar] [CrossRef] [PubMed]
- Vergnolle, N.; Bunnett, N.W.; Sharkey, K.A.; Brussee, V.; Compton, S.J.; Grady, E.F.; Cirino, G.; Gerard, N.; Basbaum, A.I.; Andrade-Gordon, P.; et al. Proteinase-activated receptor-2 and hyperalgesia: A novel pain pathway. Nat. Med. 2001, 7, 821–826. [Google Scholar] [CrossRef] [PubMed]
- White, F.A.; Jung, H.; Miller, R.J. Chemokines and the pathophysiology of neuropathic pain. Proc. Natl. Acad. Sci. USA 2007, 104, 20151–20158. [Google Scholar] [CrossRef] [PubMed]
- Cesare, P.; Dekker, L.V.; Sardini, A.; Parker, P.J.; McNaughton, P.A. Specific Involvement of PKC-ε in Sensitization of the Neuronal Response to Painful Heat. Neuron 1999, 23, 617–624. [Google Scholar] [CrossRef]
- Velazquez, K.T.; Mohammad, H.; Sweitzer, S.M. Protein kinase C in pain: Involvement of multiple isoforms. Pharmacol. Res. 2007, 55, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Baba, H.; Brenner, G.J.; Woolf, C.J. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat. Neurosci. 1999, 2, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Aley, K.O.; Martin, A.; McMahon, T.; Mok, J.; Levine, J.D.; Messing, R.O. Nociceptor Sensitization by Extracellular Signal-Regulated Kinases. J. Neurosci. 2001, 21, 6933–6939. [Google Scholar] [PubMed]
- Hu, H.J.; Bhave, G.; Gereau, R.W.T. Prostaglandin and protein kinase A-dependent modulation of vanilloid receptor function by metabotropic glutamate receptor 5: Potential mechanism for thermal hyperalgesia. J. Neurosci. 2002, 22, 7444–7452. [Google Scholar] [PubMed]
- Bhave, G.; Hu, H.-J.; Glauner, K.S.; Zhu, W.; Wang, H.; Brasier, D.J.; Oxford, G.S.; Gereau, R.W. Protein Kinase C Phosphorylation Sensitizes but Does Not Activate the Capsaicin Receptor Transient Receptor Potential Vanilloid 1 (TRPV1). Proc. Natl. Acad. Sci. USA 2003, 100, 12480–12485. [Google Scholar] [CrossRef] [PubMed]
- Morenilla-Palao, C.; Planells-Cases, R.; Garcia-Sanz, N.; Ferrer-Montiel, A. Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J. Biol. Chem. 2004, 279, 25665–25672. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.D.; Chaplan, S.R.; Higuera, E.S.; Sorkin, L.S.; Stauderman, K.A.; Williams, M.E.; Yaksh, T.L. Upregulation of Dorsal Root Ganglion alpha2delta Calcium Channel Subunit and Its Correlation with Allodynia in Spinal Nerve-Injured Rats. J. Neurosci. 2001, 21, 1868–1875. [Google Scholar] [PubMed]
- Baker, M.D. Protein kinase C mediates up-regulation of tetrodotoxin-resistant, persistent Na+ current in rat and mouse sensory neurones. J. Physiol. 2005, 567, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Karim, F.; Wang, C.C.; Gereau, R.W.T. Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J. Neurosci. 2001, 21, 3771–3779. [Google Scholar] [PubMed]
- Fundytus, M.E.; Osborne, M.G.; Henry, J.L.; Coderre, T.J.; Dray, A. Antisense oligonucleotide knockdown of mGluR1 alleviates hyperalgesia and allodynia associated with chronic inflammation. Pharmacol. Biochem. Behav. 2002, 73, 401–410. [Google Scholar] [CrossRef]
- Hu, H.J.; Alter, B.J.; Carrasquillo, Y.; Qiu, C.S.; Gereau, R.W.T. Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J. Neurosci. 2007, 27, 13181–13191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-Q.; Chiechio, S.; Sun, Y.-G.; Zhang, K.-H.; Zhao, C.-S.; Scott, M.; Johnson, R.L.; Deneris, E.S.; Renner, K.J.; Gereau, R.W.; et al. Mice Lacking Central Serotonergic Neurons Show Enhanced Inflammatory Pain and an Impaired Analgesic Response to Antidepressant Drugs. J. Neurosci. 2007, 27, 6045–6053. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, A.; Marsch, L.A.; Joseph, H.; Portenoy, R.K. Opioids and the Treatment of Chronic Pain: Controversies, Current Status, and Future Directions. Exp. Clin. Psychopharmacol. 2008, 16, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, X.; Yang, Y.; Chao, D.; Lazarus, L.H.; Xia, Y. Current research on opioid receptor function. Curr. Drug Targets 2012, 13, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Stein, C. Opioids, sensory systems and chronic pain. Eur. J. Pharmacol. 2013, 716, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Bourinet, E.; Soong, T.W.; Stea, A.; Snutch, T.P. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc. Natl. Acad. Sci. USA 1996, 93, 1486–1491. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.; Wyse, B.D.; Meutermans, W.; Smith, M.T. In vivo profiling of seven common opioids for antinociception, constipation and respiratory depression: No two opioids have the same profile. Br. J. Pharmacol. 2014, 172, 532–548. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, S.; Glanz, J.; Binswanger, I.A. National trends in pharmaceutical opioid related overdose deaths compared to other substance related overdose deaths: 1999–2009. Drug Alcohol Depend. 2013, 131, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Brady, K.T.; McCauley, J.L.; Back, S.E. Prescription Opioid Misuse, Abuse, and Treatment in the United States: An Update. Am. J. Psychiatry 2016, 173, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Backonja, M.; Beydoun, A.; Edwards, K.R.; Schwartz, S.L.; Fonseca, V.; Hes, M.; LaMoreaux, L.; Garofalo, E. Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: A randomized controlled trial. JAMA 1998, 280, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, M.; Harden, N.; Stacey, B.; Bernstein, P.; Magnus-Miller, L. Gabapentin for the treatment of postherpetic neuralgia: A randomized controlled trial. JAMA 1998, 280, 1837–1842. [Google Scholar] [CrossRef] [PubMed]
- Staats, P.S.; Yearwood, T.; Charapata, S.G.; Presley, R.W.; Wallace, M.S.; Byas-Smith, M.; Fisher, R.; Bryce, D.A.; Mangieri, E.A.; Luther, R.R.; et al. Intrathecal Ziconotide in the Treatment of Refractory Pain in Patients With Cancer or AIDS: A Randomized Controlled Trial. JAMA 2004, 291, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Rauck, R.L.; Wallace, M.S.; Leong, M.S.; Minehart, M.; Webster, L.R.; Charapata, S.G.; Abraham, J.E.; Buffington, D.E.; Ellis, D.; Kartzinel, R.; et al. A Randomized, Double-Blind, Placebo-Controlled Study of Intrathecal Ziconotide in Adults with Severe Chronic Pain. J. Pain Symptom Manag. 2006, 31, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Schmidtko, A.; Lotsch, J.; Freynhagen, R.; Geisslinger, G. Ziconotide for treatment of severe chronic pain. Lancet 2010, 375, 1569–1577. [Google Scholar] [CrossRef]
- Bell, T.J.; Thaler, C.; Castiglioni, A.J.; Helton, T.D.; Lipscombe, D. Cell-Specific Alternative Splicing Increases Calcium Channel Current Density in the Pain Pathway. Neuron 2004, 41, 127–138. [Google Scholar] [CrossRef]
- Raingo, J.; Castiglioni, A.J.; Lipscombe, D. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat. Neurosci. 2007, 10, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kremer, M.; Salvat, E.; Muller, A.; Yalcin, I.; Barrot, M. Antidepressants and gabapentinoids in neuropathic pain: Mechanistic insights. Neuroscience 2016, 338, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpaa, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.R.; Hasaballah, N.; Vetter, I. Pharmacological screening technologies for venom peptide discovery. Neuropharmacology 2017. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Hodgson, W.C.; Adams, D.J.; McIntyre, P. CHAPTER 4 Venoms-Based Drug Discovery: Bioassays, Electrophysiology, High-Throughput Screens and Target Identification. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; King, G.F., Ed.; The Royal Society of Chemistry: London, UK, 2015; pp. 97–128. [Google Scholar]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure-activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114, 901–926. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Lavergne, V.; Alewood, P.F.; Mobli, M.; King, G.F. CHAPTER 2 The Structural Universe of Disulfide-Rich Venom Peptides. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; King, G.F., Ed.; The Royal Society of Chemistry: London, UK, 2015; pp. 37–79. [Google Scholar]
- Daly, N.L.; Rosengren, K.J.; Henriques, S.T.; Craik, D.J. NMR and protein structure in drug design: Application to cyclotides and conotoxins. Eur. Biophys. J. 2011, 40, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Craik, D.J. Engineering cyclic peptide toxins. Methods Enzymol. 2012, 503, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Brady, M.R.; Baell, B.J.; Norton, S.R. Strategies for the Development of Conotoxins as New Therapeutic Leads. Mar. Drugs 2013, 11, 2293–2313. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.E.; Gavenonis, J.; Kritzer, J.A. Getting in shape: Controlling peptide bioactivity and bioavailability using conformational constraints. ACS Chem. Biol. 2013, 8, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Nareoja, K.; Nasman, J. Selective targeting of G-protein-coupled receptor subtypes with venom peptides. Acta Physiol. 2012, 204, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.; Kleinman, W.A.; Singh, L.; Singh, G.; Raufman, J.P. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem. 1992, 267, 7402–7405. [Google Scholar] [PubMed]
- Syed, Y.Y.; McCormack, P.L. Exenatide Extended-Release: An Updated Review of Its Use in Type 2 Diabetes Mellitus. Drugs 2015, 75, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Gazerani, P.; Cairns, B.E. Venom-based biotoxins as potential analgesics. Expert Rev. Neurother. 2014, 14, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Seger, J.; Horvath, M.P.; Fedosov, A.E. Prey-Capture Strategies of Fish-Hunting Cone Snails: Behavior, Neurobiology and Evolution. Brain. Behav. Evol. 2015, 86, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Jones, A.; Lewis, R.J. Remarkable inter- and intra-species complexity of conotoxins revealed by LC/MS. Peptides 2009, 30, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Bowery, N.G.; Doble, A.; Hill, D.R.; Hudson, A.L.; Shaw, J.S.; Turnbull, M.J. Baclofen: A selective agonist for a novel type of GABA receptor. Br. J. Pharmacol. 1979, 67, 444–445. [Google Scholar]
- Bowery, N.G.; Hill, D.R.; Hudson, A.L.; Doble, A.; Middlemiss, D.N.; Shaw, J.; Turnbull, M. (−)Baclofen decreases neurotransmitter release in the mammalian CNS by an action at a novel GABA receptor. Nature 1980, 283, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.R.; Bowery, N.G. 3H-baclofen and 3H-GABA bind to bicuculline-insensitive GABAB sites in rat brain. Nature 1981, 290, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Bettler, B.; Kaupmann, K.; Mosbacher, J.; Gassmann, M. Molecular Structure and Physiological Functions of GABAB Receptors. Physiol. Rev. 2004, 84, 835–867. [Google Scholar] [CrossRef] [PubMed]
- Kaupmann, K.; Huggel, K.; Heid, J.; Flor, P.J.; Bischoff, S.; Mickel, S.J.; McMaster, G.; Angst, C.; Bittiger, H.; Froestl, W.; et al. Expression cloning of GABA(B) receptors uncovers similarity to metabotropic glutamate receptors. Nature 1997, 386, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Duthey, B.; Caudron, S.; Perroy, J.; Bettler, B.; Fagni, L.; Pin, J.P.; Prezeau, L. A single subunit (GB2) is required for G-protein activation by the heterodimeric GABA(B) receptor. J. Biol. Chem. 2002, 277, 3236–3241. [Google Scholar] [CrossRef] [PubMed]
- Kniazeff, J.; Galvez, T.; Labesse, G.; Pin, J.P. No ligand binding in the GB2 subunit of the GABA(B) receptor is required for activation and allosteric interaction between the subunits. J. Neurosci. 2002, 22, 7352–7361. [Google Scholar] [PubMed]
- White, J.H.; Wise, A.; Main, M.J.; Green, A.; Fraser, N.J.; Disney, G.H.; Barnes, A.A.; Emson, P.; Foord, S.M.; Marshall, F.H. Heterodimerization is required for the formation of a functional GABAB receptor. Nature 1998, 396, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Margeta-Mitrovic, M.; Jan, Y.N.; Jan, L.Y. A trafficking checkpoint controls GABA(B) receptor heterodimerization. Neuron 2000, 27, 97–106. [Google Scholar] [CrossRef]
- Robbins, M.J.; Calver, A.R.; Filippov, A.K.; Hirst, W.D.; Russell, R.B.; Wood, M.D.; Nasir, S.; Couve, A.; Brown, D.A.; Moss, S.J.; et al. GABA(B2) is essential for g-protein coupling of the GABA(B) receptor heterodimer. J. Neurosci. 2001, 21, 8043–8052. [Google Scholar] [PubMed]
- Geng, Y.; Xiong, D.; Mosyak, L.; Malito, D.L.; Kniazeff, J.; Chen, Y.; Burmakina, S.; Quick, M.; Bush, M.; Javitch, J.A.; et al. Structure and functional interaction of the extracellular domain of human GABA(B) receptor GBR2. Nat. Neurosci. 2012, 15, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Bush, M.; Mosyak, L.; Wang, F.; Fan, Q.R. Structural mechanism of ligand activation in human GABA(B) receptor. Nature 2013, 504, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, J.; Metz, M.; Zolles, G.; Turecek, R.; Fritzius, T.; Bildl, W.; Tarusawa, E.; Kulik, A.; Unger, A.; Ivankova, K.; et al. Native GABA(B) receptors are heteromultimers with a family of auxiliary subunits. Nature 2010, 465, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Menon-Johansson, A.S.; Berrow, N.; Dolphin, A.C. Go transduces GABAB-receptor modulation of N-type calcium channels in cultured dorsal root ganglion neurons. Pflügers Arch. 1993, 425, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Mintz, I.M.; Bean, B.P. GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron 1993, 10, 889–898. [Google Scholar] [CrossRef]
- Harayama, N.; Shibuya, I.; Tanaka, K.; Kabashima, N.; Ueta, Y.; Yamashita, H. Inhibition of N- and P/Q-type calcium channels by postsynaptic GABAB receptor activation in rat supraoptic neurones. J. Physiol. 1998, 509, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Bussières, N.; El Manira, A. GABAB receptor activation inhibits N- and P/Q-type calcium channels in cultured lamprey sensory neurons. Brain Res. 1999, 847, 175–185. [Google Scholar] [CrossRef]
- Kajikawa, Y.; Saitoh, N.; Takahashi, T. GTP-binding protein βγ subunits mediate presynaptic calcium current inhibition by GABAB receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 8054–8058. [Google Scholar] [CrossRef] [PubMed]
- Chalifoux, J.R.; Carter, A.G. GABAB receptor modulation of voltage-sensitive calcium channels in spines and dendrites. J. Neurosci. 2011, 31, 4221–4232. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, C.; Jan, L.Y.; Stoffel, M.; Malenka, R.C.; Nicoll, R.A. G Protein-Coupled Inwardly Rectifying K+ Channels (GIRKs) Mediate Postsynaptic but Not Presynaptic Transmitter Actions in Hippocampal Neurons. Neuron 1997, 19, 687–695. [Google Scholar] [CrossRef]
- Kaupmann, K.; Schuler, V.; Mosbacher, J.; Bischoff, S.; Bittiger, H.; Heid, J.; Froestl, W.; Leonhard, S.; Pfaff, T.; Karschin, A.; et al. Human gamma-aminobutyric acid type B receptors are differentially expressed and regulate inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. USA 1998, 95, 14991–14996. [Google Scholar] [CrossRef] [PubMed]
- Rossi, P.; Mapelli, L.; Roggeri, L.; Gall, D.; de Kerchove d’Exaerde, A.; Schiffmann, S.N.; Taglietti, V.; D’Angelo, E. Inhibition of constitutive inward rectifier currents in cerebellar granule cells by pharmacological and synaptic activation of GABA receptors. Eur. J. Neurosci. 2006, 24, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Harrington, A.M.; Garcia-Caraballo, S.; Maddern, J.; Grundy, L.; Zhang, J.; Page, G.; Miller, P.E.; Craik, D.J.; Adams, D.J.; et al. Alpha-Conotoxin Vc1.1 inhibits human dorsal root ganglion neuroexcitability and mouse colonic nociception via GABAB receptors. Gut 2017, 66, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Bowery, N.G. GABAB receptor: A site of therapeutic benefit. Curr. Opin. Pharmacol. 2006, 6, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Malcangio, M. GABAB Receptors and pain. Neuropharmacology 2017. [Google Scholar] [CrossRef] [PubMed]
- Malcangio, M.; Ghelardini, C.; Giotti, A.; Malmberg-Aiello, P.; Bartolini, A. CGP 35348, a new GABAB antagonist, prevents antinociception and muscle-relaxant effect induced by baclofen. Br. J. Pharmacol. 1991, 103, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Schuler, V.; Lüscher, C.; Blanchet, C.; Klix, N.; Sansig, G.; Klebs, K.; Schmutz, M.; Heid, J.; Gentry, C.; Urban, L.; et al. Epilepsy, Hyperalgesia, Impaired Memory, and Loss of Pre- and Postsynaptic GABAB Responses in Mice Lacking GABAB(1). Neuron 2001, 31, 47–58. [Google Scholar] [CrossRef]
- Prosser, H.M.; Gill, C.H.; Hirst, W.D.; Grau, E.; Robbins, M.; Calver, A.; Soffin, E.M.; Farmer, C.E.; Lanneau, C.; Gray, J.; et al. Epileptogenesis and enhanced prepulse inhibition in GABA(B1)-deficient mice. Mol. Cell. Neurosci. 2001, 17, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, M.; Shaban, H.; Vigot, R.; Sansig, G.; Haller, C.; Barbieri, S.; Humeau, Y.; Schuler, V.; Muller, M.; Kinzel, B.; et al. Redistribution of GABAB(1) protein and atypical GABAB responses in GABAB(2)-deficient mice. J. Neurosci. 2004, 24, 6086–6097. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.L.; Drower, E.J. Effects of intrathecally administered THIP, baclofen and muscimol on nociceptive threshold. Eur. J. Pharmacol. 1984, 103, 121–125. [Google Scholar] [CrossRef]
- Dirig, D.M.; Yaksh, T.L. Intrathecal baclofen and muscimol, but not midazolam, are antinociceptive using the rat-formalin model. J. Pharmacol. Exp. Ther. 1995, 275, 219–227. [Google Scholar] [PubMed]
- Patel, S.; Naeem, S.; Kesingland, A.; Froestl, W.; Capogna, M.; Urban, L.; Fox, A. The effects of GABA(B) agonists and gabapentin on mechanical hyperalgesia in models of neuropathic and inflammatory pain in the rat. Pain 2001, 90, 217–226. [Google Scholar] [CrossRef]
- Smith, G.D.; Harrison, S.M.; Birch, P.J.; Elliott, P.J.; Malcangio, M.; Bowery, N.G. Increased sensitivity to the antinociceptive activity of (±)-baclofen in an animal model of chronic neuropathic, but not chronic inflammatory hyperalgesia. Neuropharmacology 1994, 33, 1103–1108. [Google Scholar] [CrossRef]
- Zemoura, K.; Ralvenius, W.T.; Malherbe, P.; Benke, D. The positive allosteric GABAB receptor modulator rac-BHFF enhances baclofen-mediated analgesia in neuropathic mice. Neuropharmacology 2016, 108, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Malcangio, M.; Bowery, N.G. Spinal cord SP release and hyperalgesia in monoarthritic rats: Involvement of the GABAB receptor system. Br. J. Pharmacol. 1994, 113, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Sokal, D.M.; Chapman, V. Inhibitory effects of spinal baclofen on spinal dorsal horn neurones in inflamed and neuropathic rats in vivo. Brain Res. 2003, 987, 67–75. [Google Scholar] [CrossRef]
- Potes, C.S.; Neto, F.L.; Castro-Lopes, J.M. Administration of baclofen, a γ-aminobutyric acid type B agonist in the thalamic ventrobasal complex, attenuates allodynia in monoarthritic rats subjected to the ankle-bend test. J. Neurosci. Res. 2006, 83, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.R.; Luque, A.; Olivera, B.M.; Barrett, J.; Cruz, L.J. Peptide toxins from Conus geographus venom. J. Biol. Chem. 1981, 256, 4734–4740. [Google Scholar] [PubMed]
- Nishiuchi, Y.; Sakakibara, S. Primary and secondary structure of conotoxin GI, a neurotoxic tridecapeptide from a marine snail. FEBS Lett. 1982, 148, 260–262. [Google Scholar] [CrossRef]
- Gray, W.R.; Rivier, J.E.; Galyean, R.; Cruz, L.J.; Olivera, B.M. Conotoxin MI. Disulfide bonding and conformational states. J. Biol. Chem. 1983, 258, 12247–12251. [Google Scholar] [PubMed]
- Quiram, P.A.; Sine, S.M. Identification of residues in the neuronal alpha7 acetylcholine receptor that confer selectivity for conotoxin ImI. J. Biol. Chem. 1998, 273, 11001–11006. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Kulak, J.M.; Cartier, G.E.; Jacobsen, R.B.; Yoshikami, D.; Olivera, B.M.; McIntosh, J.M. Alpha-conotoxin AuIB selectively blocks alpha3 beta4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J. Neurosci. 1998, 18, 8571–8579. [Google Scholar] [PubMed]
- Vincler, M.; Wittenauer, S.; Parker, R.; Ellison, M.; Olivera, B.M.; McIntosh, J.M. Molecular mechanism for analgesia involving specific antagonism of alpha9alpha10 nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 17880–17884. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: From structure to pathology. Prog. Neurobiol. 2004, 74, 363–396. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.A.; Frølund, B.; Liljefors, T.; Krogsgaard-Larsen, P. Neuronal nicotinic acetylcholine receptors: Structural revelations, target identifications, and therapeutic inspirations. J. Med. Chem. 2005, 48, 4705–4745. [Google Scholar] [CrossRef] [PubMed]
- Armishaw, C.J. Synthetic α-Conotoxin Mutants as Probes for Studying Nicotinic Acetylcholine Receptors and in the Development of Novel Drug Leads. Toxins 2010, 2, 1471–1499. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, K.E.; Peigneur, S.; Wijesekara, I.; Tytgat, J. Conotoxins Targeting Nicotinic Acetylcholine Receptors: An Overview. Mar. Drugs 2014, 12, 2970–3004. [Google Scholar] [CrossRef] [PubMed]
- Satkunanathan, N.; Livett, B.; Gayler, K.; Sandall, D.; Down, J.; Khalil, Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005, 1059, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 2010, 49, 6545–6548. [Google Scholar] [CrossRef] [PubMed]
- Klimis, H.; Adams, D.J.; Callaghan, B.; Nevin, S.; Alewood, P.F.; Vaughan, C.W.; Mozar, C.A.; Christie, M.J. A novel mechanism of inhibition of high-voltage activated calcium channels by alpha-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain 2011, 152, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Napier, I.A.; Klimis, H.; Rycroft, B.K.; Jin, A.H.; Alewood, P.F.; Motin, L.; Adams, D.J.; Christie, M.J. Intrathecal α-conotoxins Vc1.1, AuIB and MII acting on distinct nicotinic receptor subtypes reverse signs of neuropathic pain. Neuropharmacology 2012, 62, 2202–2207. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, L.; Cinci, L.; Micheli, L.; Zanardelli, M.; Pacini, A.; McIntosh, J.M.; Ghelardini, C. Alpha-conotoxin RgIA protects against the development of nerve injury-induced chronic pain and prevents both neuronal and glial derangement. Pain 2014, 155, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Pacini, A.; Micheli, L.; Maresca, M.; Branca, J.J.; McIntosh, J.M.; Ghelardini, C.; Di Cesare Mannelli, L. The alpha9alpha10 nicotinic receptor antagonist alpha-conotoxin RgIA prevents neuropathic pain induced by oxaliplatin treatment. Exp. Neurol. 2016, 282, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Romero, H.K.; Christensen, S.B.; Di Cesare Mannelli, L.; Gajewiak, J.; Ramachandra, R.; Elmslie, K.S.; Vetter, D.E.; Ghelardini, C.; Iadonato, S.P.; Mercado, J.L.; et al. Inhibition of alpha9alpha10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. USA 2017, 114, E1825–E1832. [Google Scholar] [CrossRef] [PubMed]
- Metabolic. Metabolic Discontinues Clinical Trial Programme for Neuropathic Pain Drug, ACV1. 2007. Available online: http://www.asx.com.au/asxpdf/20061129/pdf/3zv2c96tyh1nx.pdf (accessed on 17 February 2017).
- Callaghan, B.; Haythornthwaite, A.; Berecki, G.; Clark, R.J.; Craik, D.J.; Adams, D.J. Analgesic alpha-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J. Neurosci. 2008, 28, 10943–10951. [Google Scholar] [CrossRef] [PubMed]
- Daly, N.L.; Callaghan, B.; Clark, R.J.; Nevin, S.T.; Adams, D.J.; Craik, D.J. Structure and activity of alpha-conotoxin PeIA at nicotinic acetylcholine receptor subtypes and GABA(B) receptor-coupled N-type calcium channels. J. Biol. Chem. 2011, 286, 10233–10237. [Google Scholar] [CrossRef] [PubMed]
- Cuny, H.; de Faoite, A.; Huynh, T.G.; Yasuda, T.; Berecki, G.; Adams, D.J. Gamma-Aminobutyric acid type B (GABAB) receptor expression is needed for inhibition of N-type (Cav2.2) calcium channels by analgesic alpha-conotoxins. J. Biol. Chem. 2012, 287, 23948–23957. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Fischer, H.; Nevin, S.T.; Adams, D.J.; Craik, D.J. The synthesis, structural characterization, and receptor specificity of the alpha-conotoxin Vc1.1. J. Biol. Chem. 2006, 281, 23254–23263. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Yoshikami, D.; Mahe, E.; Nielsen, D.B.; Rivier, J.E.; Gray, W.R.; Olivera, B.M. A nicotinic acetylcholine receptor ligand of unique specificity, alpha-conotoxin ImI. J. Biol. Chem. 1994, 269, 16733–16739. [Google Scholar] [PubMed]
- Dolphin, A.C. G Protein Modulation of Voltage-Gated Calcium Channels. Pharmacol. Rev. 2003, 55, 607–627. [Google Scholar] [CrossRef] [PubMed]
- Berecki, G.; McArthur, J.R.; Cuny, H.; Clark, R.J.; Adams, D.J. Differential Cav2.1 and Cav2.3 channel inhibition by baclofen and alpha-conotoxin Vc1.1 via GABAB receptor activation. J. Gen. Physiol. 2014, 143, 465–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, T.G.; Cuny, H.; Slesinger, P.A.; Adams, D.J. Novel mechanism of voltage-gated N-type (Cav2.2) calcium channel inhibition revealed through alpha-conotoxin Vc1.1 activation of the GABA(B) receptor. Mol. Pharmacol. 2015, 87, 240–250. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Absalom, N.; Chebib, M.; Elgoyhen, A.B.; Vincler, M. Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem. Pharmacol. 2009, 78, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Berecki, G. Mechanisms of conotoxin inhibition of N-type (Ca(v)2.2) calcium channels. Biochim. Biophys. Acta 2013, 1828, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.B.; Hone, A.J.; Roux, I.; Kniazeff, J.; Pin, J.P.; Upert, G.; Servent, D.; Glowatzki, E.; McIntosh, J.M. RgIA4 Potently Blocks Mouse alpha9alpha10 nAChRs and Provides Long Lasting Protection against Oxaliplatin-Induced Cold Allodynia. Front. Cell. Neurosci. 2017, 11, 219. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.B.; Norimatsu, Y.; McIntosh, J.M.; Elmslie, K.S. Limited Efficacy of α-Conopeptides, Vc1.1 and RgIA, To Inhibit Sensory Neuron CaV Current. eNeuro 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Van Lierop, B.J.; Robinson, S.D.; Kompella, S.N.; Belgi, A.; McArthur, J.R.; Hung, A.; MacRaild, C.A.; Adams, D.J.; Norton, R.S.; Robinson, A.J. Dicarba alpha-conotoxin Vc1.1 analogues with differential selectivity for nicotinic acetylcholine and GABAB receptors. ACS Chem. Biol. 2013, 8, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, S.; Belgi, A.; Bartels, P.; van Lierop, B.J.; Robinson, S.D.; Kompella, S.N.; Hung, A.; Callaghan, B.P.; Adams, D.J.; Robinson, A.J.; et al. Dicarba analogues of alpha-conotoxin RgIA. Structure, stability, and activity at potential pain targets. J. Med. Chem. 2014, 57, 9933–9944. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Seymour, V.A.L.; Berecki, G.; Jia, X.; Akcan, M.; Adams, D.J.; Kaas, Q.; Craik, D.J. Less is More: Design of a Highly Stable Disulfide-Deleted Mutant of Analgesic Cyclic α-Conotoxin Vc1.1. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Carstens, B.B.; Berecki, G.; Daniel, J.T.; Lee, H.S.; Jackson, K.A.V.; Tae, H.S.; Sadeghi, M.; Castro, J.; O’Donnell, T.; Deiteren, A.; et al. Structure-Activity Studies of Cysteine-Rich α-Conotoxins that Inhibit High-Voltage-Activated Calcium Channels via GABAB Receptor Activation Reveal a Minimal Functional Motif. Angew. Chem. Int. Ed. 2016, 55, 4692–4696. [Google Scholar] [CrossRef] [PubMed]
- Nevin, S.T.; Clark, R.J.; Klimis, H.; Christie, M.J.; Craik, D.J.; Adams, D.J. Are alpha9alpha10 nicotinic acetylcholine receptors a pain target for alpha-conotoxins? Mol. Pharmacol. 2007, 72, 1406–1410. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Callaghan, B.; Berecki, G. Analgesic conotoxins: Block and G protein-coupled receptor modulation of N-type (CaV2.2) calcium channels. Br. J. Pharmacol. 2012, 166, 486–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, S.A.; Christie, M.J. Conotoxin Interactions with alpha9alpha10-nAChRs: Is the alpha9alpha10-Nicotinic Acetylcholine Receptor an Important Therapeutic Target for Pain Management? Toxins 2015, 7, 3916–3932. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; Servent, D.; McIntosh, J.M. α9-containing nicotinic acetylcholine receptors and the modulation of pain. Br. J. Pharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Townsend, A.; Livett, B.G.; Bingham, J.P.; Truong, H.T.; Karas, J.A.; O’Donnell, P.; Williamson, N.A.; Purcell, A.W.; Scanlon, D. Mass Spectral Identification of Vc1.1 and Differential Distribution of Conopeptides in the Venom Duct of Conus victoriae. Effect of Post-Translational Modifications and Disulfide Isomerisation on Bioactivity. Int. J. Pept. Res. Ther. 2009, 15, 195–203. [Google Scholar] [CrossRef]
- Kalinichev, M.; Donovan-Rodriguez, T.; Girard, F.; Riguet, E. Evaluation of peripheral versus central effects of GABAB receptor activation using a novel, positive allosteric modulator of the GABAB receptor ADX71943, a pharmacological tool compound with a fully peripheral activity profile. Br. J. Pharmacol. 2014, 171, 4941–4954. [Google Scholar] [CrossRef] [PubMed]
- Kalinichev, M.; Donovan-Rodriguez, T.; Girard, F.; Haddouk, H.; Royer-Urios, I.; Schneider, M.; Bate, S.T.; Marker, C.; Pomonis, J.D.; Poli, S. ADX71943 and ADX71441, novel positive allosteric modulators of the GABAB receptor with distinct central/peripheral profiles, show efficacy in the monosodium iodoacetate model of chronic osteoarthritis pain in the rat. Eur. J. Pharmacol. 2017, 795, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Christie, M.J. α9-Nicotinic Acetylcholine Receptors Contribute to the Maintenance of Chronic Mechanical Hyperalgesia, but Not Thermal or Mechanical Allodynia. Mol. Pain 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Holtman, J.R.; Dwoskin, L.P.; Dowell, C.; Wala, E.P.; Zhang, Z.; Crooks, P.A.; McIntosh, J.M. The novel small molecule alpha9alpha10 nicotinic acetylcholine receptor antagonist ZZ-204G is analgesic. Eur. J. Pharmacol. 2011, 670, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Wala, E.P.; Crooks, P.A.; McIntosh, J.M.; Holtman, J.R., Jr. Novel small molecule alpha9alpha10 nicotinic receptor antagonist prevents and reverses chemotherapy-evoked neuropathic pain in rats. Anesth. Analg. 2012, 115, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.G.; Norberg, T.; Griffin, D.; Hoeger, C.; Akhtar, M.; Schmidt, K.; Low, W.; Dykert, J.; Richelson, E.; Navarro, V.; et al. Contulakin-G, an O-glycosylated invertebrate neurotensin. J. Biol. Chem. 1999, 274, 13752–13759. [Google Scholar] [CrossRef] [PubMed]
- White, J.F.; Noinaj, N.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; Tate, C.G.; et al. Structure of the agonist-bound neurotensin receptor. Nature 2012, 490, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Boules, M.; Li, Z.; Smith, K.; Fredrickson, P.; Richelson, E. Diverse Roles of Neurotensin Agonists in the Central Nervous System. Front. Endocrinol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Kleczkowska, P.; Lipkowski, A.W. Neurotensin and neurotensin receptors: Characteristic, structure-activity relationship and pain modulation—A review. Eur. J. Pharmacol. 2013, 716, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.P.; Wang, J.; Dong, Y.L.; Wang, Y.Y.; Li, Y.Q. The roles of neurotensin and its analogues in pain. Curr. Pharm. Des. 2015, 21, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Carraway, R.E.; Dobner, P.R. Endogenous neurotensin facilitates visceral nociception and is required for stress-induced antinociception in mice and rats. Neuroscience 2004, 126, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Roussy, G.; Dansereau, M.A.; Dore-Savard, L.; Belleville, K.; Beaudet, N.; Richelson, E.; Sarret, P. Spinal NTS1 receptors regulate nociceptive signaling in a rat formalin tonic pain model. J. Neurochem. 2008, 105, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Guillemette, A.; Dansereau, M.A.; Beaudet, N.; Richelson, E.; Sarret, P. Intrathecal administration of NTS1 agonists reverses nociceptive behaviors in a rat model of neuropathic pain. Eur. J. Pain 2012, 16, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.W.; Hofer, K.; McCumber, D.; Wagstaff, J.D.; Layer, R.T.; McCabe, R.T.; Yaksh, T.L. An assessment of the antinociceptive efficacy of intrathecal and epidural contulakin-G in rats and dogs. Anesth. Analg. 2007, 104, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.E.; Allen, J.; Wagstaff, J.; Shafer, S.L.; Yaksh, T. The pharmacokinetics of the conopeptide contulakin-G (CGX-1160) after intrathecal administration: An analysis of data from studies in beagles. Anesth. Analg. 2007, 104, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Zhang, L.; Smith, M.D.; Walewska, A.; Vellore, N.A.; Baron, R.; McIntosh, J.M.; White, H.S.; Olivera, B.M.; Bulaj, G. A marine analgesic peptide, Contulakin-G, and neurotensin are distinct agonists for neurotensin receptors: Uncovering structural determinants of desensitization properties. Front. Pharmacol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Sang, C.N.; Barnabe, K.J.; Kern, S.E. Phase IA Clinical Trial Evaluating the Tolerability, Pharmacokinetics, and Analgesic Efficacy of an Intrathecally Administered Neurotensin A Analogue in Central Neuropathic Pain Following Spinal Cord Injury. Clin. Pharmacol. Drug Dev. 2016, 5, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Vanderah, T.W. Delta and kappa opioid receptors as suitable drug targets for pain. Clin. J. Pain 2010, 26, S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Kivell, B.; Prisinzano, T.E. Kappa opioids and the modulation of pain. Psychopharmacology 2010, 210, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Butelman, E.R.; Yuferov, V.; Kreek, M.J. Kappa opioid receptor/dynorphin system: Genetic and pharmacotherapeutic implications for addiction. Trends Neurosci. 2012, 35, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Binder, W.; Walker, J.S. Effect of the peripherally selective kappa-opioid agonist, asimadoline, on adjuvant arthritis. Br. J. Pharmacol. 1998, 124, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Bileviciute-Ljungar, I.; Saxne, T.; Spetea, M. Anti-inflammatory effects of contralateral administration of the kappa-opioid agonist U-50,488H in rats with unilaterally induced adjuvant arthritis. Rheumatology 2006, 45, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Schepers, R.J.; Mahoney, J.L.; Gehrke, B.J.; Shippenberg, T.S. Endogenous kappa-opioid receptor systems inhibit hyperalgesia associated with localized peripheral inflammation. Pain 2008, 138, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Auh, Q.S.; Ro, J.Y. Effects of peripheral κ opioid receptor activation on inflammatory mechanical hyperalgesia in male and female rats. Neurosci. Lett. 2012, 524, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Desmeules, J.A.; Kayser, V.; Guilbaud, G. Selective opioid receptor agonists modulate mechanical allodynia in an animal model of neuropathic pain. Pain 1993, 53, 277–285. [Google Scholar] [CrossRef]
- Keita, H.; Kayser, V.; Guilbaud, G. Antinociceptive effect of a kappa-opioid receptor agonist that minimally crosses the blood-brain barrier (ICI 204448) in a rat model of mononeuropathy. Eur. J. Pharmacol. 1995, 277, 275–280. [Google Scholar] [CrossRef]
- Eliav, E.; Herzberg, U.; Caudle, R.M. The kappa opioid agonist GR89,696 blocks hyperalgesia and allodynia in rat models of peripheral neuritis and neuropathy. Pain 1999, 79, 255–264. [Google Scholar] [CrossRef]
- Rivière, P.J.M. Peripheral kappa-opioid agonists for visceral pain. Br. J. Pharmacol. 2004, 141, 1331–1334. [Google Scholar] [CrossRef] [PubMed]
- Brust, A.; Croker, D.E.; Colless, B.; Ragnarsson, L.; Andersson, A.; Jain, K.; Garcia-Caraballo, S.; Castro, J.; Brierley, S.M.; Alewood, P.F.; et al. Conopeptide-Derived kappa-Opioid Agonists (Conorphins): Potent, Selective, and Metabolic Stable Dynorphin A Mimetics with Antinociceptive Properties. J. Med. Chem. 2016, 59, 2381–2395. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.A.; Castro, J.; Harrington, A.M.; Isaacs, N.; Moretta, M.; Hicks, G.A.; Urso, D.M.; Brierley, S.M. Increased kappa-opioid receptor expression and function during chronic visceral hypersensitivity. Gut 2014, 63, 1199–1200. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Whately, E.; Brust, A.; Inserra, M.C.; Asvadi, N.H.; Lewis, R.J.; Alewood, P.F.; Cabot, P.J.; Vetter, I. Activation of κ Opioid Receptors in Cutaneous Nerve Endings by Conorphin-1, a Novel Subtype-Selective Conopeptide, Does Not Mediate Peripheral Analgesia. ACS Chem. Neurosci. 2015, 6, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.; Jolkkonen, M.; Mulugeta, E.; Onali, P.; Adem, A. Snake toxins with high selectivity for subtypes of muscarinic acetylcholine receptors. Biochimie 2000, 82, 793–806. [Google Scholar] [CrossRef]
- Ellis, J.L.; Harman, D.; Gonzalez, J.; Spera, M.L.; Liu, R.; Shen, T.Y.; Wypij, D.M.; Zuo, F. Development of muscarinic analgesics derived from epibatidine: Role of the M4 receptor subtype. J. Pharmacol. Exp. Ther. 1999, 288, 1143–1150. [Google Scholar] [PubMed]
- Harvey, A.L.; Kornisiuk, E.; Bradley, K.N.; Cervenansky, C.; Duran, R.; Adrover, M.; Sanchez, G.; Jerusalinsky, D. Effects of muscarinic toxins MT1 and MT2 from green mamba on different muscarinic cholinoceptors. Neurochem. Res. 2002, 27, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Duttaroy, A.; Gomeza, J.; Gan, J.W.; Siddiqui, N.; Basile, A.S.; Harman, W.D.; Smith, P.L.; Felder, C.C.; Levey, A.I.; Wess, J. Evaluation of muscarinic agonist-induced analgesia in muscarinic acetylcholine receptor knockout mice. Mol. Pharmacol. 2002, 62, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.R.; Pan, H.L. Up-regulation of spinal muscarinic receptors and increased antinociceptive effect of intrathecal muscarine in diabetic rats. J. Pharmacol. Exp. Ther. 2003, 307, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.W.; Martino, G.; Coupal, M.; Lindberg, M.; Schroeder, P.; Santhakumar, V.; Valiquette, M.; Sandin, J.; Widzowski, D.; Laird, J. Broad analgesic activity of a novel, selective M1 agonist. Neuropharmacology 2017, 123, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, I.A.; Thomas, L.; Loughnan, M.; Motin, L.; Palant, E.; Croker, D.E.; Alewood, D.; Chen, S.; Graham, R.M.; Alewood, P.F.; et al. Allosteric alpha 1-adrenoreceptor antagonism by the conopeptide rho-TIA. J. Biol. Chem. 2003, 278, 34451–34457. [Google Scholar] [CrossRef] [PubMed]
- Quinton, L.; Girard, E.; Maiga, A.; Rekik, M.; Lluel, P.; Masuyer, G.; Larregola, M.; Marquer, C.; Ciolek, J.; Magnin, T.; et al. Isolation and pharmacological characterization of AdTx1, a natural peptide displaying specific insurmountable antagonism of the alpha1A-adrenoceptor. Br. J. Pharmacol. 2010, 159, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Rouget, C.; Quinton, L.; Maïga, A.; Gales, C.; Masuyer, G.; Malosse, C.; Chamot-Rooke, J.; Thai, R.; Mourier, G.; De Pauw, E.; et al. Identification of a novel snake peptide toxin displaying high affinity and antagonist behaviour for the α2-adrenoceptors. Br. J. Pharmacol. 2010, 161, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, G.; Collet, G.; Mourier, G.; Gilles, N.; Fruchart-Gaillard, C.; Marcon, E.; Servent, D. Polypharmacology profiles and phylogenetic analysis of three-finger toxins from mamba venom: Case of aminergic toxins. Biochimie 2014, 103, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; de Santos, V.; Zafaralla, G.C.; Ramilo, C.A.; Zeikus, R.; Gray, W.R.; Olivera, B.M. Invertebrate vasopressin/oxytocin homologs. Characterization of peptides from Conus geographus and Conus straitus venoms. J. Biol. Chem. 1987, 262, 15821–15824. [Google Scholar] [PubMed]
- Dutertre, S.; Croker, D.; Daly, N.L.; Andersson, A.; Muttenthaler, M.; Lumsden, N.G.; Craik, D.J.; Alewood, P.F.; Guillon, G.; Lewis, R.J. Conopressin-T from Conus tulipa reveals an antagonist switch in vasopressin-like peptides. J. Biol. Chem. 2008, 283, 7100–7108. [Google Scholar] [CrossRef] [PubMed]
- González-Hernández, A.; Rojas-Piloni, G.; Condés-Lara, M. Oxytocin and analgesia: Future trends. Trends Pharmacol. Sci. 2014, 35, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Kabli, N.; Cahill, C.M. Anti-allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain 2007, 127, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Junyan, X.; Zhiwei, W.; Xiuli, Z.; Xinmiao, L.; Olivier, C. BmK-YA, an enkephalin-like peptide in scorpion venom. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Bardoni, R.; Tawfik, V.L.; Wang, D.; Francois, A.; Solorzano, C.; Shuster, S.A.; Choudhury, P.; Betelli, C.; Cassidy, C.; Smith, K.; et al. Delta opioid receptors presynaptically regulate cutaneous mechanosensory neuron input to the spinal cord dorsal horn. Neuron 2014, 81, 1312–1327. [Google Scholar] [CrossRef] [PubMed]
- Ducancel, F. The sarafotoxins. Toxicon 2002, 40, 1541–1545. [Google Scholar] [CrossRef]
- Smith, T.P.; Haymond, T.; Smith, S.N.; Sweitzer, S.M. Evidence for the endothelin system as an emerging therapeutic target for the treatment of chronic pain. J. Pain Res. 2014, 7, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Maillo, M.; Aguilar, M.B.; Lopez-Vera, E.; Craig, A.G.; Bulaj, G.; Olivera, B.M.; Heimer de la Cotera, E.P. Conorfamide, a Conus venom peptide belonging to the RFamide family of neuropeptides. Toxicon 2002, 40, 401–407. [Google Scholar] [CrossRef]
- Ayachi, S.; Simonin, F. Involvement of Mammalian RF-Amide Peptides and Their Receptors in the Modulation of Nociception in Rodents. Front. Endocrinol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Safavi-Hemami, H.; Raghuraman, S.; Imperial, J.S.; Papenfuss, A.T.; Teichert, R.W.; Purcell, A.W.; Olivera, B.M.; Norton, R.S. Discovery by proteogenomics and characterization of an RF-amide neuropeptide from cone snail venom. J. Proteom. 2015, 114, 38–47. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Peptide | Species | Pharmacological Target | Indication | Year Approved |
|---|---|---|---|---|
| Captopril | Brazilian pit viper (Bothrops jararaca) | Angiotensin-converting enzyme inhibitor | Hypertension | 1981 |
| Eptifibatide | Southeastern pygmy rattlesnake (Sistrurus miliarius barbouri) | Platelet glycoprotein IIb/IIIa receptor inhibitor | Unstable angina | 1998 |
| MVIIA (Ziconotide) | Cone snail (Conus magus) | CaV2.2 inhibitor | Neuropathic pain | 2004 |
| Exenatide | Gila monster (Heloderma suspectum) | Insulin secretagogue | Type 2 diabetes mellitus | 2005 |
| α-Conotoxin | Sequence a | nAChR Selectivity | Cav2.2 IC50 (nM) b | Analgesic Activity | References |
|---|---|---|---|---|---|
| Vc1.1 | GCCSDPRCNYDHPEIC | α9α10 > α3β2 ~ α3β4 | 1.7 | PNL, CCI, CVH | [122,129,138] |
| RgIA | GCCSDPRCRYRCR | α9α10 > α3β2 ~ α3β4 | 7.3 | PNL, CCI, chemotherapy | [122] |
| Vc1a | GCCSDORCNYDHPγIC | α9α10 | Inactive | Inactive | [122,136] |
| ImI | GCCSDPRCAWRC | α7 | Inactive | N.D. | [135,139] |
| PeIA | GCCSHPACSVNHPELC | α9α10 ~ α3β2 | 1.1 | N.D. | [136] |
| AuIB | GCCSYPPCFATNPDC | α3β4 | 1.5 | PNL, CCI | [129] |
| MII | GCCSNPVCHLEHSNLC | α3β2 | Inactive | PNL | [129] |
| GPCR Family | Venom Peptide Ligands | Subtype Selectivity (Gα Subunit) | Evidence for Analgesic Role | References |
|---|---|---|---|---|
| Muscarinic acetylcholine receptor | MT1 MT2 MT4 MT5 MT7 | M1 (Gαq) | Expressed throughout peripheral nociceptive and central nerves and are dysregulated in pain conditions.
Various subtype selective and non-selective mAChR agonists are analgesic in rodent models of pain. | [73,187,188,189,190,191,192] |
| MT3 MT6 | M4 (Gαi) | |||
| MTLP-1 | M3 (Gαq) | |||
| α-Adrenergic receptor | ρ-Da1a ρ-TIA | α1A (Gαq) α1B (Gαq) | Widely expressed on nociceptors and central pathways and mediate descending inhibition. Several α2-selective agonists exhibit analgesic properties in rodents | [193,194,195,196] |
| ρ-Da1b MTα MT1 | α2A (Gαi) α2A α2B (Gαi) | |||
| MT3 | α1B ~ α2A | |||
| Oxytocin/vasopressin receptor | Conopressin-G Conopressin-S Conopressin-T | OTR (Gαq) OTR V1a (Gαq) > OTR | Oxytocin acting via OTR and V1a is analgesic in animal models of pain. | [197,198,199] |
| Opioid receptor | BmK-YA | δOR (Gαi) | Involved in presynaptic control of nociceptive inputs onto dorsal horn.
Administration of δ-opioid agonists produces anti-allodynia in chronic pain models. | [173,200,201,202] |
| Endothelin receptor | SRTX-a SRTX-b SRTX-c | ETA, ETB (Gαq) | Distributed in central and peripheral pathways. Implicated as a regulator of acute and chronic pain. | [203,204] |
| S6c | ETB > ETA | |||
| Neuropeptide FF receptor | CNF-Sr1 CNF-Sr2 Vc1 | None (Gαi) | Important role in modulating pain signalling in the CNS. | [205,206,207] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daniel, J.T.; Clark, R.J. G-Protein Coupled Receptors Targeted by Analgesic Venom Peptides. Toxins 2017, 9, 372. https://doi.org/10.3390/toxins9110372
Daniel JT, Clark RJ. G-Protein Coupled Receptors Targeted by Analgesic Venom Peptides. Toxins. 2017; 9(11):372. https://doi.org/10.3390/toxins9110372
Chicago/Turabian StyleDaniel, James T., and Richard J. Clark. 2017. "G-Protein Coupled Receptors Targeted by Analgesic Venom Peptides" Toxins 9, no. 11: 372. https://doi.org/10.3390/toxins9110372
APA StyleDaniel, J. T., & Clark, R. J. (2017). G-Protein Coupled Receptors Targeted by Analgesic Venom Peptides. Toxins, 9(11), 372. https://doi.org/10.3390/toxins9110372