Development and Validation of a Liquid Chromatography-Tandem Mass Spectrometry Method Coupled with Dispersive Solid-Phase Extraction for Simultaneous Quantification of Eight Paralytic Shellfish Poisoning Toxins in Shellfish

Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of HPLC-MS/MS Conditions
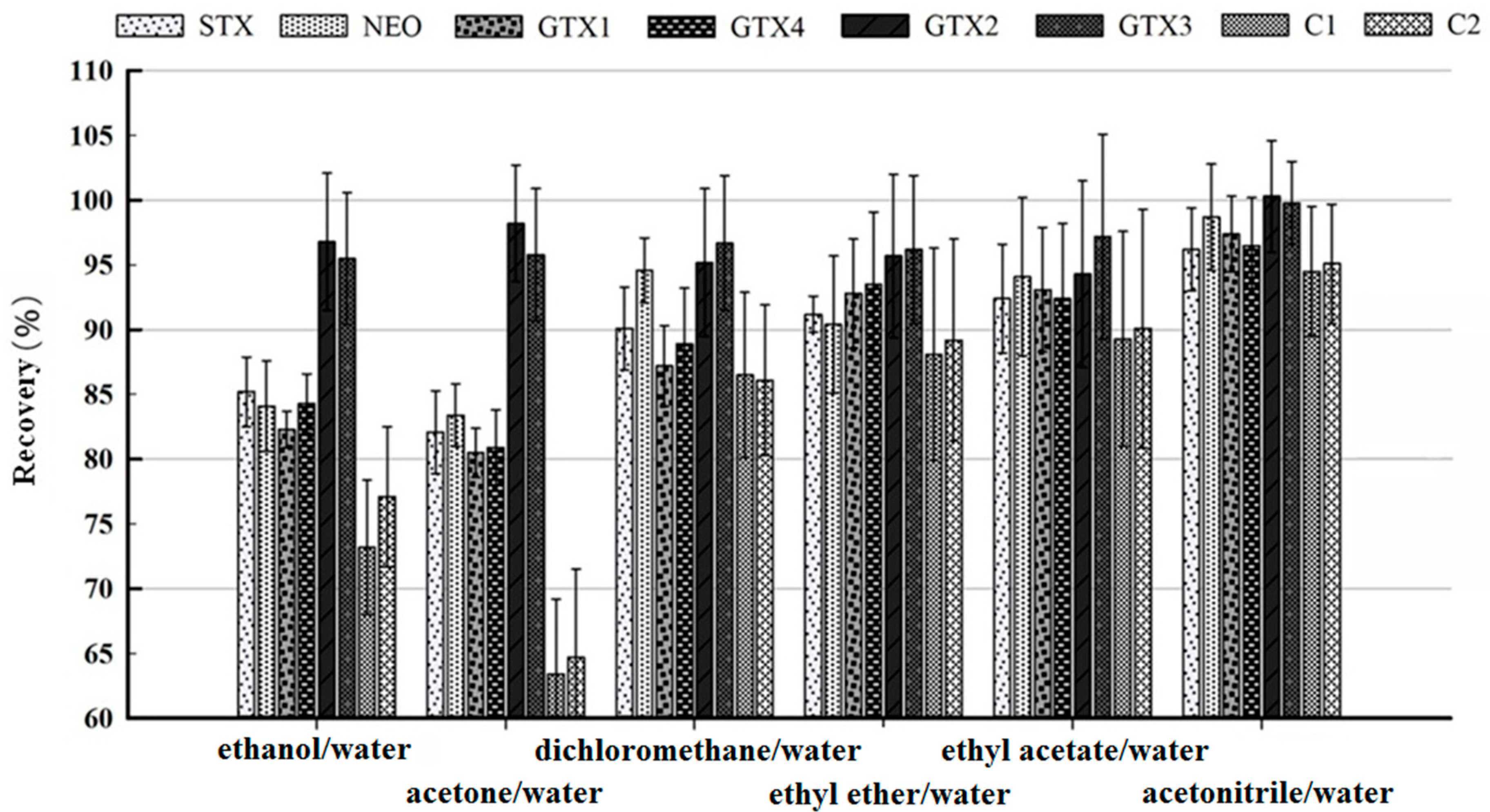
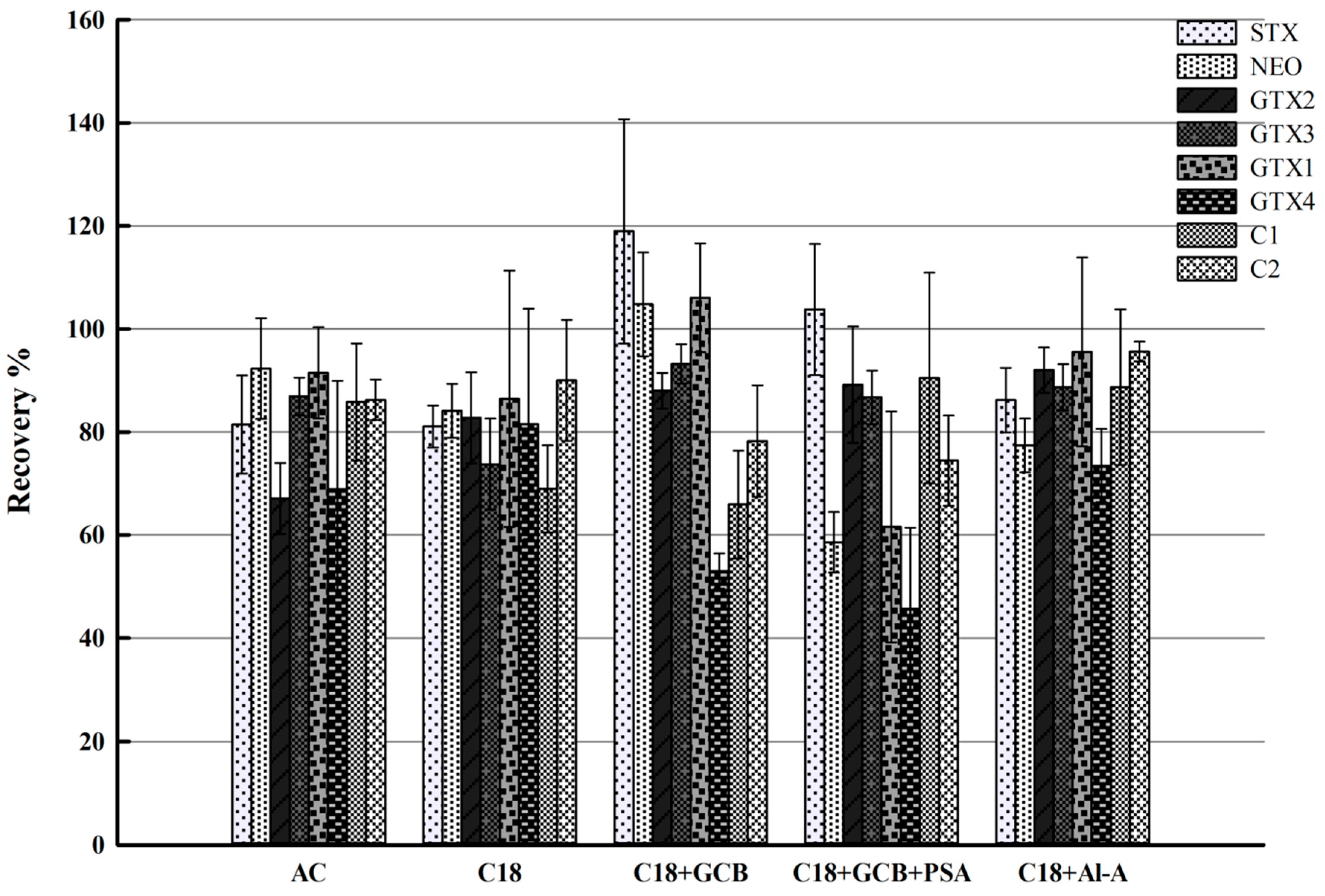
2.2. Optimization of Sample Pretreatment
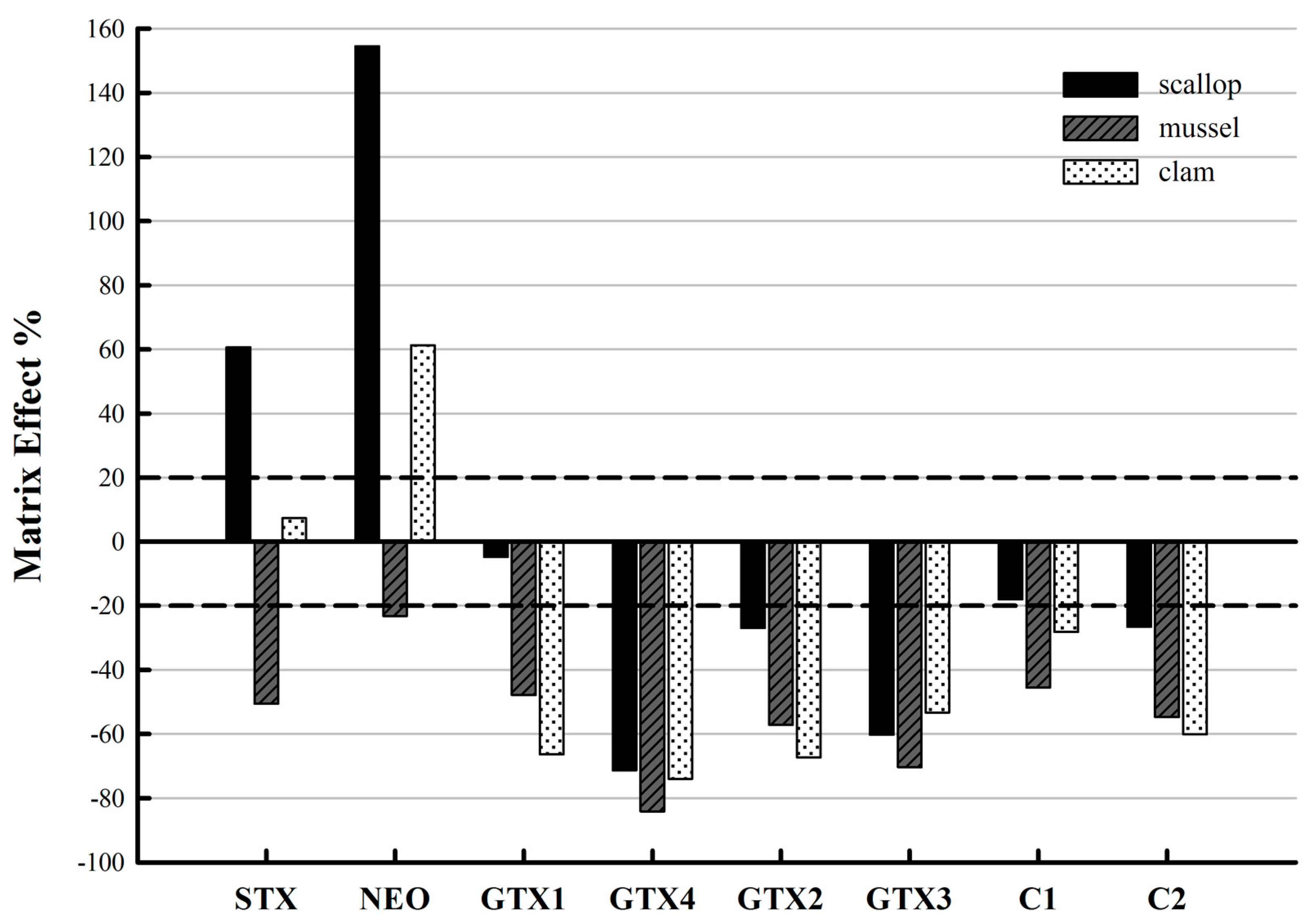
2.3. Evaluation of the Matrix Effects
2.4. Method Validation
2.5. Sample Analysis
3. Conclusions
4. Materials and Methods
4.1. Materials and Reagents
4.2. Preparation of Standard Solutions
4.3. Samples
4.4. Sample Pretreatment
4.5. LC-MS/MS Analysis
4.6. Evaluation of Matrix Effects
4.7. Validation of the Method
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hall, S.; Strichartz, G.; Moczydlowski, E.; Ravindran, A.; Reichardt, P.B. The saxitoxins Sources, Chemistry, and Pharmacology. In Marine Toxins Origin, Structure, and Molecular Pharmacology; Hall, S., Strichartz, G., Eds.; American Chemical Society: Washington, DC, USA, 1990; Volume 418, pp. 29–65. [Google Scholar]
- Shimizu, Y. Microalgal metabolites. Chem. Rev. 1993, 93, 1685–1698. [Google Scholar] [CrossRef]
- Anderson, D.M.; Cembella, A.D.; Hallegraeff, G.M. Progress in understanding harmful algal blooms: Paradigm shifts and new technologies for research, monitoring, and management. Annu. Rev. Mar. Sci. 2012, 4, 143–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z. Neurotoxins from marine dinoflagellates: A brief review. Mar. Drugs 2008, 6, 349–371. [Google Scholar] [CrossRef] [PubMed]
- Faber, S. Saxitoxin and the induction of paralytic shellfish poisoning. J. Young Investig. 2012, 23, 1–7. [Google Scholar]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y. Postcolumn derivatization liquid-chromatographic method for paralytic shellfish toxins. J. AOAC Int. 1995, 78, 528–532. [Google Scholar]
- Wekell, J.C.; Hurst, J.; Lefebvre, K.A. The origin of the regulatory limits for psp and asp toxins in shellfish. J. Shellfish Res. 2004, 23, 927–930. [Google Scholar]
- Toyofuku, H. Joint fao/who/ioc activities to provide scientific advice on marine biotoxins (research report). Mar. Pollut. Bull. 2006, 52, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Benford, D.; Cockburn, A.; Cravedi, J.; Dogliotti, E.; Di Domenico, A.; Fernández-Cruz, M.; Fink-Gremmels, J.; Fürst, P.; Galli, C. Scientific opinion of the panel on contaminants in the food chain on a request from the european commission on marine biotoxins in shellfish—Saxitoxin group. EFSA J. 2009, 1019, 1–76. [Google Scholar]
- Hellrich, K. Paralytic Shellfish Poison; Association of Official Analytical Chemists: Arlington, VA, USA, 1990; pp. 881–882. [Google Scholar]
- Commission Regulation. Commission Regulation (EC) No 1664/2006 of 6 November 2006 Amending Regulation (EC) No 2074/2005 as Regards Implementing Measures for Certain Products of Animal Origin Intended for Human Consumption and Repealing Certain Implementing Measures; Off J Eur Union: Brussels, Belgium, 2006; pp. 13–45. [Google Scholar]
- Asp, T.N.; Larsen, S.; Aune, T. Analysis of psp toxins in norwegian mussels by a post-column derivatization hplc method. Toxicon 2004, 43, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.J.; Wekell, M.M. Determination of paralytic shellfish poisoning toxins by high-pressure liquid-chromatography. ACS Symp. Ser. 1984, 262, 197–205. [Google Scholar]
- Lawrence, J.F.; Niedzwiadek, B. Quantitative determination of paralytic shellfish poisoning toxins in shellfish by using prechromatographic oxidation and liquid chromatography with fluorescence detection. J. AOAC Int. 2001, 84, 1099–1108. [Google Scholar] [PubMed]
- Lawrence, J.; Ménard, C. Liquid chromatographic determination of paralytic shellfish poisons in shellfish after prechromatographic oxidation. J. Assoc. Off. Anal. Chem. 1991, 74, 1006–1012. [Google Scholar] [PubMed]
- Lawrence, J.F.; Niedzwiadek, B.; Menard, C. Quantitative determination of paralytic shellfish poisoning toxins in shellfish using prechromatographic oxidation and liquid chromatography with fluorescence detection: Collaborative study. J. AOAC Int. 2005, 88, 1714–1732. [Google Scholar] [PubMed]
- DeGrasse, S.L.; van de Riet, J.; Hatfield, R.; Turner, A. Pre-versus post-column oxidation liquid chromatography fluorescence detection of paralytic shellfish toxins. Toxicon 2011, 57, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Rourke, W.A.; Murphy, C.J.; Pitcher, G.; van de Riet, J.M.; Burns, B.G.; Thomas, K.M.; Quilliam, M.A. Rapid postcolumn methodology for determination of paralytic shellfish toxins in shellfish tissue. J. AOAC Int. 2008, 91, 589–597. [Google Scholar] [PubMed]
- Harju, K.; Rapinoja, M.L.; Avondet, M.A.; Arnold, W.; Schär, M.; Luginbühl, W.; Kremp, A.; Suikkanen, S.; Kankaanpää, H.; Burrell, S. Results of a saxitoxin proficiency test including characterization of reference material and stability studies. Toxins 2015, 7, 4852–4867. [Google Scholar] [CrossRef] [PubMed]
- Dell’Aversano, C.; Eaglesham, G.K.; Quilliam, M.A. Analysis of cyanobacterial toxins by hydrophilic interaction liquid chromatography-mass spectrometry. J. Chromatogr. A 2004, 1028, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Dell’Aversano, C.; Hess, P.; Quilliam, M.A. Hydrophilic interaction liquid chromatography–mass spectrometry for the analysis of paralytic shellfish poisoning (psp) toxins. J. Chromatogr. A 2005, 2, 190–201. [Google Scholar] [CrossRef]
- Mattarozzi, M.; Milioli, M.; Bianchi, F.; Cavazza, A.; Pigozzi, S.; Milandri, A.; Careri, M. Optimization of a rapid quechers sample treatment method for hilic-ms 2 analysis of paralytic shellfish poisoning (psp) toxins in mussels. Food Control 2016, 60, 138–145. [Google Scholar] [CrossRef]
- Turrell, E.; Stobo, L.; Lacaze, J.P.; Piletsky, S.; Piletska, E. Optimization of hydrophilic interaction liquid chromatography/mass spectrometry and development of solid-phase extraction for the determination of paralytic shellfish poisoning toxins. J. AOAC Int. 2008, 91, 1372–1386. [Google Scholar] [PubMed]
- Boundy, M.J.; Selwood, A.I.; Harwood, D.T.; McNabb, P.S.; Turner, A.D. Development of a sensitive and selective liquid chromatography-mass spectrometry method for high throughput analysis of paralytic shellfish toxins using graphitised carbon solid phase extraction. J. Chromatogr. A 2015, 1387, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Diener, M.; Erler, K.; Christian, B.; Luckas, B. Application of a new zwitterionic hydrophillic interaction chromatography column for determination of paralytic shellfish poisoning toxins. J. Sep. Sci. 2007, 30, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Matsushima, R.; Harada, T.; Oikawa, H.; Murata, M.; Suzuki, T. Quantitative determination of paralytic shellfish toxins in cultured toxic algae by lc-ms/ms. Food Addit. Contam. A 2013, 30, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Halme, M.; Rapinoja, M.L.; Karjalainen, M.; Vanninen, P. Verification and quantification of saxitoxin from algal samples using fast and validated hydrophilic interaction liquid chromatography-tandem mass spectrometry method. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 880, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Jansson, D.; Åstot, C. Analysis of paralytic shellfish toxins, potential chemical threat agents, in food using hydrophilic interaction liquid chromatography–mass spectrometry. J. Chromatogr. A 2015, 1417, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, L.Y.; Yin, Y.C.; Fu, W.S.; Qiu, B.; Lin, Z.Y.; Yang, Y.Q.; Zheng, L.M.; Li, J.R.; Chen, G.N. Determination of paralytic shellfish poisoning toxins by hilic-ms/ms coupled with dispersive solid phase extraction. Food Chem. 2013, 137, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Mattarozzi, M.; Bianchi, F.; Milioli, M.; Cavazza, A.; Careri, M. An innovative method based on quick, easy, cheap, effective, rugged, and safe extraction coupled to desorption electrospray ionization-high resolution mass spectrometry for screening the presence of paralytic shellfish toxins in clams. Talanta 2016, 147, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Amado, M.; Prieto-Blanco, M.C.; Lopez-Mahia, P.; Muniategui-Lorenzo, S.; Prada-Rodriguez, D. Strengths and weaknesses of in-tube solid-phase microextraction: A scoping review. Anal. Chim. Acta 2016, 906, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Anastassiades, M.; Lehotay, S.J.; Stajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [PubMed]
- Regueiro, J.; Álvarez, G.; Mauriz, A.; Blanco, J. High throughput analysis of amnesic shellfish poisoning toxins in bivalve molluscs by dispersive solid-phase extraction and high-performance liquid chromatography using a monolithic column. Food Chem. 2011, 127, 1884–1891. [Google Scholar] [CrossRef]
- Mol, H.G.J.; Plaza-Bolaños, P.; Zomer, P.; de Rijk, T.C.; Stolker, A.A.M.; Mulder, P.P.J. Toward a generic extraction method for simultaneous determination of pesticides, mycotoxins, plant toxins, and veterinary drugs in feed and food matrixes. Anal. Chem. 2008, 80, 9450–9459. [Google Scholar] [CrossRef] [PubMed]
- Gage, P.W. Tetrodotoxin and saxitoxin as pharmacological tools. In Neuropoisons: Their Pathophysiological Actions; Simpson, L.L., Ed.; Springer: Boston, MA, USA, 1971; pp. 187–212. [Google Scholar]
- Frenich, A.G.; Romero-González, R.; Gómez-Pérez, M.L.; Vidal, J.L.M. Multi-mycotoxin analysis in eggs using a quechers-based extraction procedure and ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry. J. Chromatogr. A 2011, 1218, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- European Union. Commission Decision of 14 august 2002 Implementing Council Directive 96/23/ec. Concerning the Performance of Analytical Methods and the Interpretation of Results (2002/657/ec); Off J Eur Union: Brussels, Belgium, 2002. [Google Scholar]
- Kong, F.Z.; Xu, Z.J.; Yu, R.C.; Li, Q.L. Paralytic shellfish poison(psp) monitoring and analysis in the bohai and yellow seas. Period. Ocean Univ. China 2007, 37, 305–309. [Google Scholar]

| Toxins | R1 | R2 | R3 | R4 |
|---|---|---|---|---|
| STX | H | H | H | OCONH2 |
| NEO | OH | H | H | OCONH2 |
| GTX1 | OH | H | OSO3 | OCONH2 |
| GTX2 | H | H | OSO3 | OCONH2 |
| GTX3 | H | OSO3 | H | OCONH2 |
| GTX4 | OH | OSO3 | H | OCONH2 |
| C1 | H | H | OSO3 | OCONHSO3 |
| C2 | H | OSO3 | H | OCONHSO3 |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PSP Toxins | Transition Made from | Retention Time (min) | Precursor Ion (m/z) | Product Ion (m/z) | CE (eV) |
|---|---|---|---|---|---|
| C1 | [M + H-SO3]+ | 8.38 | 396.1 | 316.1 * | 13.0 |
| 298.1 | 19.0 | ||||
| C2 | [M + H-SO3]+ | 8.97 | 396.1 | 298.1 * | 19.0 |
| 316.1 | 13.0 | ||||
| GTX2 | [M + H-SO3]+ | 9.68 | 316.1 | 220.0 * | 26.0 |
| 147.9 | 24.0 | ||||
| GTX1 | [M + H-SO3]+ | 9.75 | 332.1 | 236.1 * | 29.0 |
| 164.0 | 32.0 | ||||
| GTX3 | [M + H]+ | 10.23 | 396.1 | 298.1 * | 19.0 |
| 316.1 | 13.0 | ||||
| GTX4 | [M + H]+ | 10.33 | 412.2 | 314.2 * | 22.0 |
| 332.2 | 21.0 | ||||
| STX | [M + H]+ | 13.18 | 300.1 | 204.0 * | 25.0 |
| 282.1 | 19.0 | ||||
| NEO | [M + H]+ | 13.48 | 316.1 | 220.1 * | 21.0 |
| 298.1 | 18.0 |
| PSP Toxins | Linearity Range (ng·mL−1) | Scallop | Mussel | Clam | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R2 | LOD | LOQ | R2 | LOD | LOQ | R2 | LOD | LOQ | ||
| STX | 9.92~158.75 | 0.9982 | 0.33 | 1.32 | 0.9995 | 1.65 | 4.96 | 0.9986 | 0.82 | 2.48 |
| NEO | 10.34~165.47 | 0.9983 | 0.69 | 2.07 | 0.9982 | 2.59 | 5.17 | 0.9985 | 2.07 | 4.14 |
| GTX1 | 12.42~198.74 | 0.9991 | 4.14 | 8.28 | 0.9960 | 5.52 | 8.28 | 0.9971 | 5.52 | 8.28 |
| GTX2 | 22.57~361.15 | 0.9991 | 2.01 | 9.03 | 0.9959 | 2.82 | 11.29 | 0.9977 | 2.01 | 9.03 |
| GTX3 | 9.58~153.25 | 0.9999 | 2.55 | 6.39 | 0.9937 | 4.79 | 9.58 | 0.9996 | 1.60 | 4.79 |
| GTX4 | 4.05~64.82 | 0.9994 | 2.70 | 4.05 | 0.9957 | 3.04 | 6.08 | 0.9966 | 2.70 | 4.05 |
| C1 | 26.96~431.28 | 0.9954 | 2.27 | 9.08 | 0.9977 | 3.33 | 9.98 | 0.9994 | 2.85 | 9.98 |
| C2 | 8.06~128.93 | 0.9972 | 1.35 | 2.02 | 0.9969 | 1.61 | 3.22 | 0.9962 | 2.69 | 4.03 |
| PSP Toxins | Spike Conc. (μg kg−1) | Scallop | Mussel | Clam | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intra-Day (%) | Inter-Day (%) | Intra-Day (%) | Inter-Day (%) | Intra-Day (%) | Inter-Day (%) | ||||||||
| Recovery | RSD | Recovery | RSD | Recovery | RSD | Recovery | RSD | Recovery | RSD | Recovery | RSD | ||
| STX | 49.61 | 96.78 | 10.27 | 93.25 | 2.28 | 98.34 | 6.43 | 96.19 | 0.63 | 95.56 | 5.65 | 94.58 | 1.53 |
| 26.46 | 89.74 | 9.76 | 100.30 | 5.37 | 92.43 | 5.06 | 96.73 | 4.22 | 97.80 | 6.18 | 95.86 | 0.35 | |
| 9.92 | 98.77 | 5.39 | 95.13 | 2.88 | 92.21 | 2.80 | 94.62 | 4.35 | 85.89 | 7.76 | 89.01 | 5.47 | |
| NEO | 51.71 | 88.70 | 9.23 | 92.25 | 7.21 | 84.04 | 6.70 | 92.29 | 7.31 | 93.11 | 10.95 | 101.12 | 14.77 |
| 27.58 | 90.54 | 14.63 | 91.84 | 14.39 | 88.92 | 12.01 | 83.62 | 6.22 | 90.93 | 3.63 | 94.33 | 0.56 | |
| 10.34 | 102.96 | 9.23 | 97.41 | 3.98 | 102.13 | 13.48 | 92.20 | 3.07 | 91.41 | 3.18 | 99.29 | 4.64 | |
| GTX1 | 62.11 | 84.69 | 14.34 | 89.96 | 5.09 | 99.56 | 9.86 | 96.50 | 3.45 | 96.01 | 12.53 | 97.80 | 9.45 |
| 33.12 | 85.59 | 12.01 | 90.98 | 12.63 | 91.02 | 12.50 | 93.48 | 9.19 | 80.22 | 1.17 | 90.12 | 1.38 | |
| 12.42 | 89.18 | 18.17 | 96.42 | 4.05 | 88.28 | 16.19 | 97.87 | 2.42 | 89.32 | 19.10 | 87.66 | 0.62 | |
| GTX2 | 112.86 | 94.10 | 4.89 | 87.04 | 3.35 | 91.38 | 8.93 | 94.50 | 7.02 | 100.76 | 4.52 | 97.68 | 11.77 |
| 60.19 | 95.32 | 16.22 | 89.71 | 3.22 | 99.78 | 7.20 | 96.46 | 1.61 | 116.53 | 7.32 | 85.87 | 13.25 | |
| 22.57 | 101.78 | 2.75 | 88.47 | 12.44 | 100.08 | 5.41 | 92.88 | 18.37 | 82.22 | 14.26 | 94.50 | 5.27 | |
| GTX3 | 47.89 | 96.18 | 2.24 | 90.08 | 3.88 | 82.80 | 5.62 | 94.74 | 2.74 | 99.39 | 1.50 | 90.38 | 1.12 |
| 25.54 | 90.88 | 6.57 | 98.80 | 6.90 | 96.96 | 12.29 | 98.02 | 0.59 | 100.06 | 11.07 | 94.22 | 2.67 | |
| 9.58 | 92.36 | 8.62 | 93.96 | 17.47 | 86.36 | 5.05 | 97.04 | 0.59 | 93.60 | 13.00 | 99.01 | 6.32 | |
| GTX4 | 20.25 | 81.52 | 14.57 | 95.34 | 4.07 | 93.45 | 17.85 | 86.76 | 4.34 | 91.79 | 8.25 | 96.80 | 7.09 |
| 10.80 | 83.66 | 12.66 | 89.00 | 7.72 | 82.63 | 12.51 | 95.46 | 8.85 | 95.84 | 6.83 | 95.16 | 1.74 | |
| 4.05 | 96.90 | 11.83 | 93.84 | 7.22 | 91.57 | 5.27 | 96.93 | 5.63 | 90.19 | 5.27 | 86.40 | 6.68 | |
| C1 | 134.78 | 89.06 | 13.85 | 93.98 | 2.90 | 90.19 | 11.26 | 94.37 | 2.42 | 107.86 | 9.87 | 98.09 | 1.13 |
| 71.88 | 99.69 | 5.31 | 90.92 | 16.92 | 98.49 | 10.07 | 101.09 | 3.48 | 94.68 | 16.59 | 98.42 | 9.33 | |
| 26.96 | 94.82 | 10.19 | 97.59 | 11.21 | 96.91 | 11.23 | 88.36 | 7.38 | 86.42 | 3.20 | 94.23 | 1.69 | |
| C2 | 40.29 | 91.57 | 12.77 | 88.07 | 1.08 | 82.27 | 8.57 | 99.64 | 1.99 | 102.35 | 7.33 | 92.53 | 7.78 |
| 21.49 | 96.45 | 18.70 | 86.50 | 9.04 | 104.50 | 15.53 | 100.67 | 1.71 | 96.18 | 8.07 | 96.78 | 8.27 | |
| 8.06 | 97.10 | 2.96 | 104.68 | 11.59 | 90.34 | 6.96 | 94.41 | 10.59 | 93.40 | 17.49 | 94.80 | 4.52 | |
| Shellfish | Sample No. | Origin | STX | NEO | GTX1 | GTX2 | GTX3 | GTX4 | C1 | C2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Scallop | 1 | Dalian | - * | - | - | - | - | - | - | - |
| 2 | Qinhuangdao | - | - | - | - | - | - | - | - | |
| 3 | Tianjin | - | - | - | - | - | - | - | - | |
| 4 | Yantai | - | - | - | - | 16.35 | - | - | - | |
| 5 | Dalian | - | - | - | - | - | - | - | - | |
| 6 | Quanzhou | - | - | - | - | - | - | - | - | |
| 7 | Ningbo | - | - | - | 7.85 | - | - | - | - | |
| 8 | Qingdao | - | - | - | - | - | 12.31 | - | - | |
| 9 | Rizhao | - | - | - | - | - | - | - | - | |
| 10 | Xiamen | - | - | - | - | - | - | 16.32 | - | |
| Clam | 11 | Lianyungang | - | - | - | - | 6.24 | - | - | - |
| 12 | Dalian | - | - | - | - | - | - | - | - | |
| 13 | Qinhuangdao | - | - | - | - | - | - | - | - | |
| 14 | Tianjin | - | - | - | - | - | - | - | - | |
| 15 | Yantai | - | - | - | - | - | - | - | - | |
| 16 | Dalian | - | - | - | - | - | - | - | - | |
| 17 | Quanzhou | - | - | - | - | - | - | - | - | |
| 18 | Ningbo | - | - | - | - | 15.68 | - | - | - | |
| 19 | Qingdao | - | - | - | - | - | - | - | - | |
| 20 | Rizhao | - | - | - | - | - | - | - | - | |
| Mussel | 21 | Ningbo | - | - | - | - | - | - | - | - |
| 22 | Qingdao | - | - | - | - | 11.44 | 20.82 | - | - | |
| 23 | Rizhao | - | - | - | - | 6.34 | - | - | - | |
| 24 | Quanzhou | - | - | - | - | - | - | - | - | |
| 25 | Qinhuangdao | - | - | - | - | - | - | - | - | |
| 26 | Tianjin | - | - | - | - | 9.87 | - | - | - | |
| 27 | Yantai | - | - | - | - | - | - | - | - | |
| 28 | Taizhou | - | - | - | - | - | 18.91 | - | - | |
| 29 | Quanzhou | - | - | 9.67 | - | - | - | - | - | |
| 30 | Dalian | - | - | - | - | - | - | - | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Zhou, L.; Tan, Y.; Shi, X.; Zhao, Z.; Nie, D.; Zhou, C.; Liu, H. Development and Validation of a Liquid Chromatography-Tandem Mass Spectrometry Method Coupled with Dispersive Solid-Phase Extraction for Simultaneous Quantification of Eight Paralytic Shellfish Poisoning Toxins in Shellfish. Toxins 2017, 9, 206. https://doi.org/10.3390/toxins9070206
Yang X, Zhou L, Tan Y, Shi X, Zhao Z, Nie D, Zhou C, Liu H. Development and Validation of a Liquid Chromatography-Tandem Mass Spectrometry Method Coupled with Dispersive Solid-Phase Extraction for Simultaneous Quantification of Eight Paralytic Shellfish Poisoning Toxins in Shellfish. Toxins. 2017; 9(7):206. https://doi.org/10.3390/toxins9070206
Chicago/Turabian StyleYang, Xianli, Lei Zhou, Yanglan Tan, Xizhi Shi, Zhiyong Zhao, Dongxia Nie, Changyan Zhou, and Hong Liu. 2017. "Development and Validation of a Liquid Chromatography-Tandem Mass Spectrometry Method Coupled with Dispersive Solid-Phase Extraction for Simultaneous Quantification of Eight Paralytic Shellfish Poisoning Toxins in Shellfish" Toxins 9, no. 7: 206. https://doi.org/10.3390/toxins9070206
APA StyleYang, X., Zhou, L., Tan, Y., Shi, X., Zhao, Z., Nie, D., Zhou, C., & Liu, H. (2017). Development and Validation of a Liquid Chromatography-Tandem Mass Spectrometry Method Coupled with Dispersive Solid-Phase Extraction for Simultaneous Quantification of Eight Paralytic Shellfish Poisoning Toxins in Shellfish. Toxins, 9(7), 206. https://doi.org/10.3390/toxins9070206