
Phage Display Analysis of Monoclonal Antibody Binding to Anthrax Toxin Lethal Factor


Abstract
:1. Introduction
2. Results
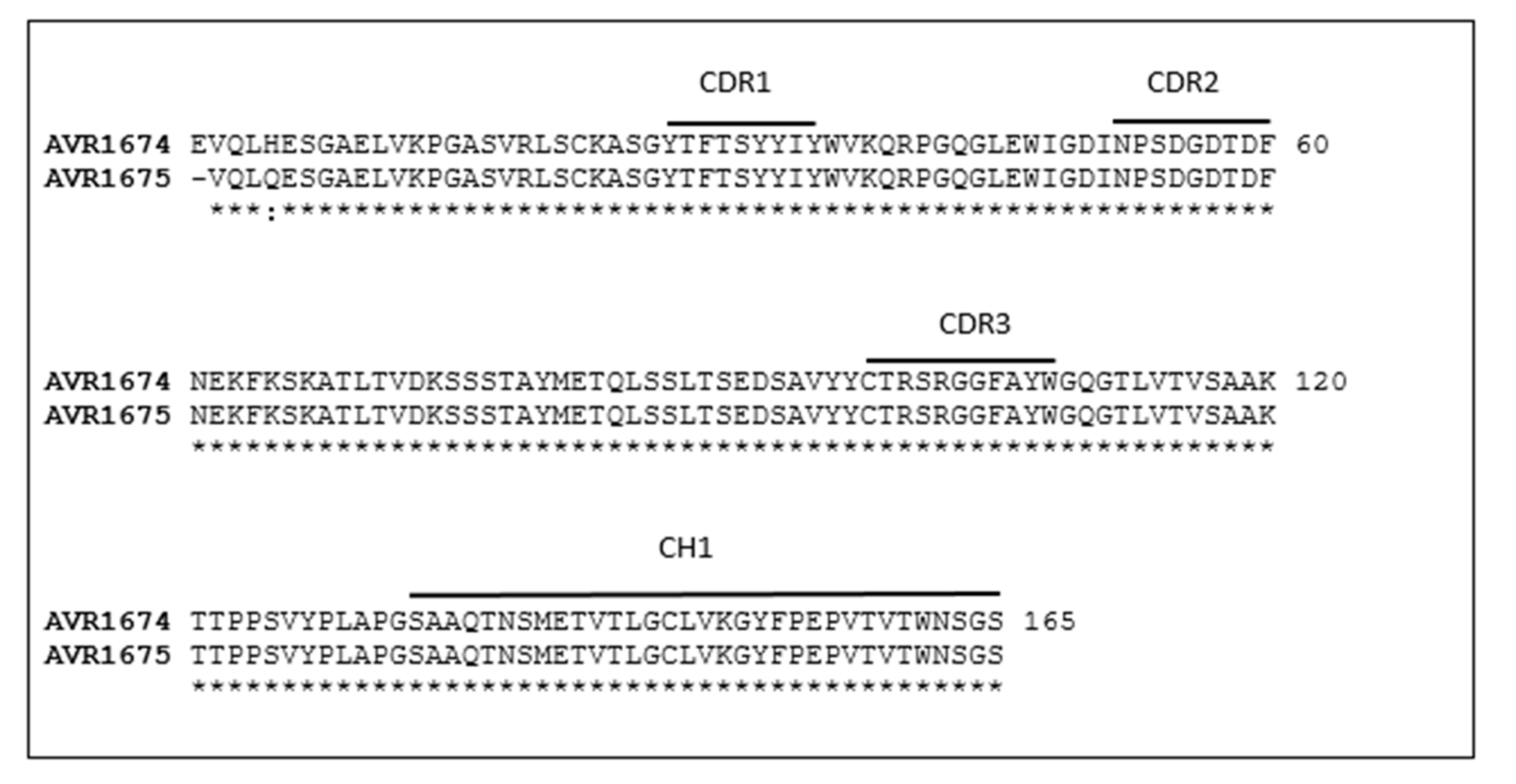
2.1. Comparative Analysis of AVR1674 and AVR1675 mAb Sequences
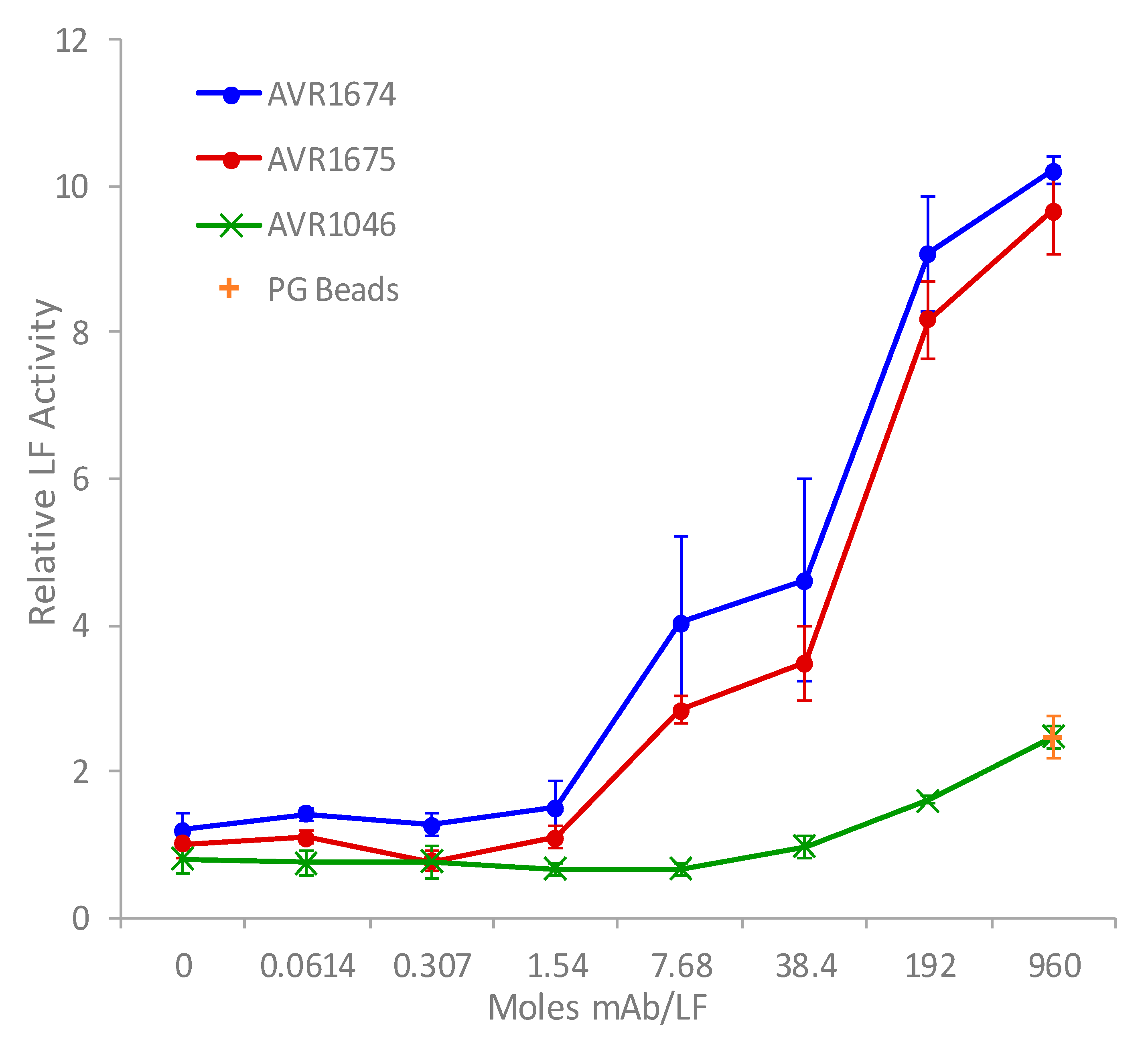
2.2. Antibody-Enhanced Activity of rLF In Vitro
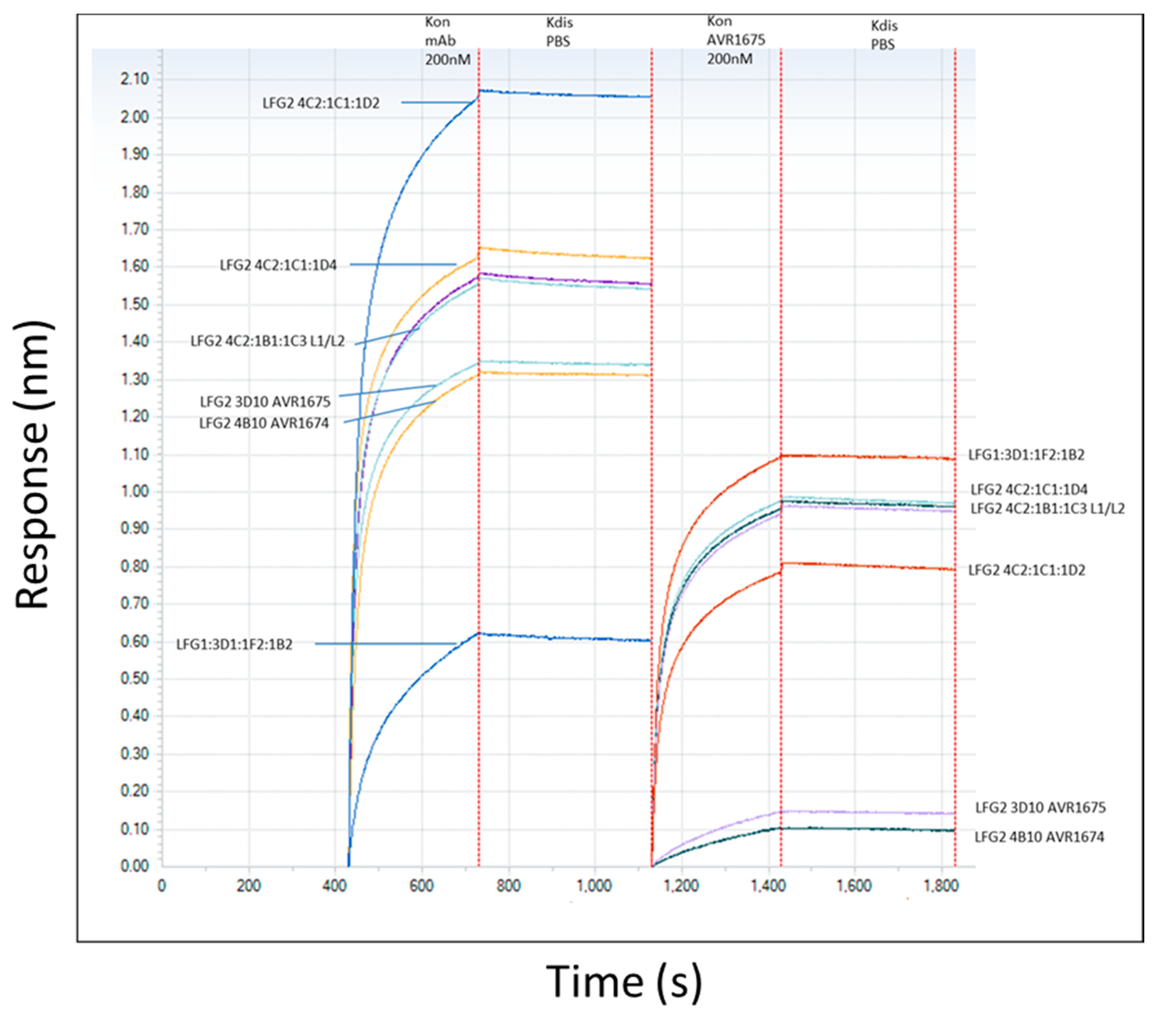
2.3. Competitive Binding of AVR1674 and AVR1675 to rLF
2.4. Identification and Sequence Analysis of mAb-Binding Phage Peptide Sequences
2.5. Analysis of Synthetic Oligopeptide Binding to mAb
3. Discussion
4. Materials and Methods
4.1. Protein Reagents
4.2. Development of mAbs AVR1674 and AVR1675
4.3. Sequence Analysis of mAbs AVR1674 and AVR1675
4.4. Isotope-Dilution MALDI-TOF MS Quantification of mAb Enhanced LF Activity
4.5. Label-Free Binding and Competition Analyses
4.6. Binding Site Mapping and Competition with Phage Display Peptides
4.7. Phage Sequencing and Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mock, M.; Fouet, A. Anthrax. Annu. Rev. Microbiol. 2001, 55, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Brossier, F.; Mock, M. Toxins of Bacillus anthracis. Toxicon 2001, 39, 1747–1755. [Google Scholar] [CrossRef]
- Klimpel, K.R.; Arora, N.; Leppla, S.H. Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol. Microbiol. 1994, 13, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Janowiak, B.E.; Brzozowski, M.; Juryck, J.; Falke, S.; Gogol, E.P.; Collier, R.J.; Fisher, M.T. GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat. Struct. Mol. Biol. 2008, 15, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar] [CrossRef]
- Collier, R.J. Mechanism of membrane translocation by anthrax toxin: Insertion and pore formation by protective antigen. J. Appl. Microbiol. 1999, 87, 283. [Google Scholar] [CrossRef]
- Boyer, A.E.; Gallegos-Candela, M.; Lins, R.C.; Kuklenyik, Z.; Woolfitt, A.; Moura, H.; Kalb, S.; Quinn, C.P.; Barr, J.R. Quantitative mass spectrometry for bacterial protein toxins—A sensitive, specific, high-throughput tool for detection and diagnosis. Molecules 2011, 16, 2391–2413. [Google Scholar] [CrossRef]
- Boyer, A.E.; Quinn, C.P.; Woolfitt, A.R.; Pirkle, J.L.; McWilliams, L.G.; Stamey, K.L.; Bagarozzi, D.A.; Hart J.C., Jr.; Barr, J.R. Detection and quantification of anthrax lethal factor in serum by mass spectrometry. Anal. Chem. 2007, 79, 8463–8470. [Google Scholar] [CrossRef]
- Stoddard, R.A.; Quinn, C.P.; Schiffer, J.M.; Boyer, A.E.; Goldstein, J.; Bagarozzi, D.A.; Soroka, S.D.; Dauphin, L.A.; Hoffimaster, A.R. Detection of anthrax protective antigen (PA) using europium labeled anti-PA monoclonal antibody and time-resolved fluorescence. J. Immunol. Methods 2014, 408, 78–88. [Google Scholar] [CrossRef]
- Boyer, A.E.; Quinn, C.P.; Beesley, C.A.; Gallegos-Candela, M.; Marston, C.K.; Cronin, L.X.; Lins, R.C.; Stoddard, R.A.; Li, H.; Schiffer, J.; et al. Lethal factor toxemia and anti-protective antigen antibody activity in naturally acquired cutaneous anthrax. J. Infect. Dis. 2011, 204, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Nolen, L.D.; Sun, J.; Booth, M.; Donaldson, L.; Quinn, C.P.; Boyer, A.E.; Hendricks, K.; Shadomy, S.; Bothma, P.; et al. Analysis of Anthrax Immune Globulin Intravenous with Antimicrobial Treatment in Injection Drug Users, Scotland, 2009–2010. Emerg. Infect. Dis. 2017, 23, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Marston, C.K.; Ibrahim, H.; Lee, P.; Churchwell, G.; Gumke, M.; Stanek, D.; Gee, J.E.; Boyer, A.E.; Gallegos-Candela, M.; Barr, J.R.; et al. Anthrax Toxin-Expressing Bacillus cereus Isolated from an Anthrax-Like Eschar. PLoS ONE 2016, 11, e0156987. [Google Scholar] [CrossRef] [PubMed]
- Sprenkle, M.D.; Griffith, J.; Marinelli, W.; Boyer, A.E.; Quinn, C.P.; Pesik, N.T.; Hoffmaster, A.; Keenan, J.; Juni, B.A.; Blaney, D.D. Lethal factor and anti-protective antigen IgG levels associated with inhalation anthrax, Minnesota, USA. Emerg. Infect. Dis. 2014, 20, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Lim, N.K.; Kim, J.H.; Oh, M.S.; Kee, S.; Kim, K.S.; Kang, H.J.; Inn, K.S. An anthrax lethal factor-neutralizing monoclonal antibody protects rats before and after challenge with anthrax toxin. Infect. Immun. 2005, 73, 6547–6551. [Google Scholar] [CrossRef] [PubMed]
- Hoess, R.H.; Mack, A.J.; Walton, H.; Reilly, T.M. Identification of a structural epitope by using a peptide library displayed on filamentous bacteriophage. J. Immunol. (Baltim. Md. 1950) 1994, 153, 724–729. [Google Scholar]
- Bonnycastle, L.L.; Mehroke, J.S.; Rashed, M.; Gong, X.; Scott, J.K. Probing the basis of antibody reactivity with a panel of constrained peptide libraries displayed by filamentous phage. J. Mol. Biol. 1996, 258, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Felici, F.; Luzzago, A.; Folgori, A.; Cortese, R. Mimicking of discontinuous epitopes by phage-displayed peptides, II. Selection of clones recognized by a protective monoclonal antibody against the Bordetella pertussis toxin from phage peptide libraries. Gene 1993, 128, 21–27. [Google Scholar] [CrossRef]
- Schellekens, G.A.; Lasonder, E.; Feijlbrief, M.; Koedijl, D.G.; Drijfhout, J.W.; Scheffer, A.J.; Welling-Wester, S.; Welling, G.W. Identification of the core residues of the epitope of a monoclonal antibody raised against glycoprotein D of herpes simplex virus type 1 by screening of a random peptide library. Eur. J. Immunol. 1994, 24, 3188–3193. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Schopp, C.; Scott, J.K. The Performance of Epitope Libraries: Crystallographic Evidence for a Direct Correspondence between the Residues Selected and the Critical Residues for Binding. Protein Eng. 1993, 6, 106. [Google Scholar]
- Pannifer, A.D.; Wong, T.Y.; Schwarzenbacher, R.; Renatus, M.; Petosa, C.; Bienkowska, J.; Lacy, D.B.; Collier, R.J.; Parks, S.; Leppla, S.H.; et al. Crystal structure of the anthrax lethal factor. Nature 2001, 414, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Maize, K.M.; Kurbanov, E.K.; De La Mora-Rey, T.; Geders, T.W.; Hwang, D.J.; Walters, M.A.; Johnson, R.L.; Amin, E.A.; Finzel, B.C. Anthrax toxin lethal factor domain 3 is highly mobile and responsive to ligand binding. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 2813–2822. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous phage assembly: Morphogenetically defective mutants that do not kill the host. Virology 1988, 167, 156–165. [Google Scholar] [CrossRef]
- Smith, G.P.; Scott, J.K. Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol. 1993, 217, 228–257. [Google Scholar] [PubMed]
- Barbas, C.F., 3rd; Burton, D.R.; Scott, J.K.; Silverman, G.J. (Eds.) Phage Display: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Sayle, R.A.; Milner-White, E.J. RASMOL: Biomolecular graphics for all. Trends Biochem. Sci. 1995, 20, 374. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mAb Clone ID | Antibody ID | Concentration (nM) | Response (R) | KD (M) | Kassoc (1/Ms) | Correlation Coefficient (r2) |
|---|---|---|---|---|---|---|
| LFG2:4B10 | AVR1674 | 200 | 1.31 | 18.7 × 10−9 | 1.37 × 105 | 0.913 |
| LFG2:3D10 | AVR1675 | 200 | 1.34 | 17.7 × 10−9 | 1.68 × 105 | 0.875 |
| Panning Antibody Identifier | Panning Antibody Reactivity | Phage Sequence Identifier | Core Amino Acid Sequence | Phage ELISA Reactivity (High = OD > 1.0, Intermediate = OD 0.5–1.0, Low = OD < 0.5) |
|---|---|---|---|---|
| AVR1046 | Anti-PA | Control-φC1 | n h h y s h l | Negative control (OD 0.07) |
| Control-φC3 | l p l t p l p | Negative control (OD 0.05) | ||
| Control-φC4 | s p e a r h p | Negative control (OD 0.04) | ||
| Control-φC5 | y a i l e d h | Negative control (OD 0.05) | ||
| AVR1675 | Anti-LF | φC8 | t f k d e i v | High (OD 1.71) |
| φD12 | t f k d e i v | High (OD 1.31) | ||
| φE4 | t f k d e i v | High (OD 1.30) | ||
| φE11 | t f k d e i v | High (OD 1.59) | ||
| φC9 | t f k d d i h | High (OD 1.33) | ||
| φB5 | t y k d d i r | High (OD 1.65) | ||
| φD2 | t y k d d i r | High (OD 1.78) | ||
| φC7 | t f k d d l f | High (OD 1.56) | ||
| φD4 | t f k d d g y | Low (OD 0.08) | ||
| φB6 | t y l d d l y | High (OD 1.77) | ||
| φD11 | t y l d d l y | High (OD 1.70) | ||
| φE2 | t f l d d a p | High (OD 1.22) | ||
| φD8 | t w r d d i p | Intermediate (OD 0.79) | ||
| φE5 | t y r d d p p | High (OD 1.55) | ||
| φC2 | t v l d d v a | Low (OD 0.36) | ||
| φD7 | t v r d d q i | High (OD 1.78) | ||
| φB9 | t f r d e p m | High (OD 1.71) | ||
| φE12 | t v r d e p l | High (OD 1.70) | ||
| Consensus | t x k/r d e z n | N/A | ||
| | . . | | : : | ||||
| LF(644–650) | t h q d e i y | N/A |
| Peptide Descriptor | Peptide Sequence Position | Response (nm) | % R of Consensus | KD (M) * |
|---|---|---|---|---|
| 1-2-3-4-5-6-7-8-9-S-Kbio | ||||
| φC8-derived Reference Sequence | T-F-K-D-E-I-G-G-G-S-Kbio | 4.8387 | 100 | 13.9 × 10−9 |
| P1 Substitution A | A-F-K-D-E-I-G-G-G-S-Kbio | 0.5352 | 11 | 35.1 × 10−9 |
| P2 Substitution A | T-A-K-D-E-I-G-G-G-S-Kbio | 3.7854 | 78 | 31.5 × 10−9 |
| P3 Substitution A | T-F-A-D-E-I-G-G-G-S-Kbio | 0.4475 | 9 | 25.2 × 10−9 |
| P4 Substitution A | T-F-K-A-E-I-G-G-G-S-Kbio | 0.2612 | 5 | >100 × 10−9 |
| P5 Substitution A | T-F-K-D-A-I-G-G-G-S-Kbio | 1.0132 | 21 | 24.5 × 10−9 |
| P6 Substitution A | T-F-K-D-E-A-G-G-G-S-Kbio | 5.2909 | 109 | 16.4 × 10−9 |
| P2 Substitution H | T-H-K-D-E-I-G-G-G-S-Kbio | 2.3704 | 48 | 60.1 × 10−9 |
| P3 Substitution Q | T-F-Q-D-E-I-G-G-G-S-Kbio | 1.3414 | 27 | 34.4 × 10−9 |
| Native LF core sequence | T-H-Q-D-E-I-Y-E-Q-Kbio (LF644–652) | 0.7024 | 14 | >100 × 10−9 |
| Native LF extended sequence | P-N-I-A-E-Q-Y-T-H-Q-D-E-I-Y-E-Q-V-H-S-K-G-L-Y-V-P-E-S-R-G-G-G-S-Kbio (LF637–664) | 0.7024 | 14 | >100 × 10−9 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldstein, J.M.; Lee, J.; Tang, X.; Boyer, A.E.; Barr, J.R.; Bagarozzi Jr., D.A.; Quinn, C.P. Phage Display Analysis of Monoclonal Antibody Binding to Anthrax Toxin Lethal Factor. Toxins 2017, 9, 221. https://doi.org/10.3390/toxins9070221
Goldstein JM, Lee J, Tang X, Boyer AE, Barr JR, Bagarozzi Jr. DA, Quinn CP. Phage Display Analysis of Monoclonal Antibody Binding to Anthrax Toxin Lethal Factor. Toxins. 2017; 9(7):221. https://doi.org/10.3390/toxins9070221
Chicago/Turabian StyleGoldstein, Jason M., Joo Lee, Xiaoling Tang, Anne E. Boyer, John R. Barr, Dennis A. Bagarozzi Jr., and Conrad P. Quinn. 2017. "Phage Display Analysis of Monoclonal Antibody Binding to Anthrax Toxin Lethal Factor" Toxins 9, no. 7: 221. https://doi.org/10.3390/toxins9070221
APA StyleGoldstein, J. M., Lee, J., Tang, X., Boyer, A. E., Barr, J. R., Bagarozzi Jr., D. A., & Quinn, C. P. (2017). Phage Display Analysis of Monoclonal Antibody Binding to Anthrax Toxin Lethal Factor. Toxins, 9(7), 221. https://doi.org/10.3390/toxins9070221