
Lengths of the C-Terminus and Interconnecting Loops Impact Stability of Spider-Derived Gating Modifier Toxins



Abstract
:
1. Introduction
2. Results
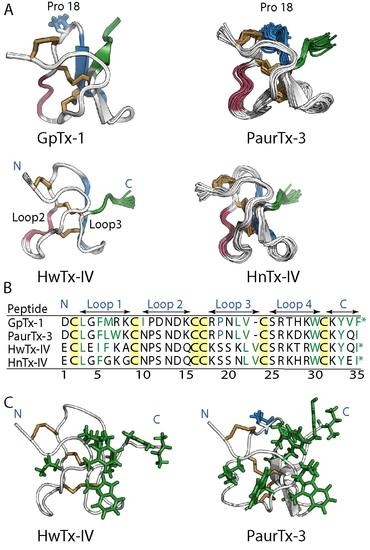
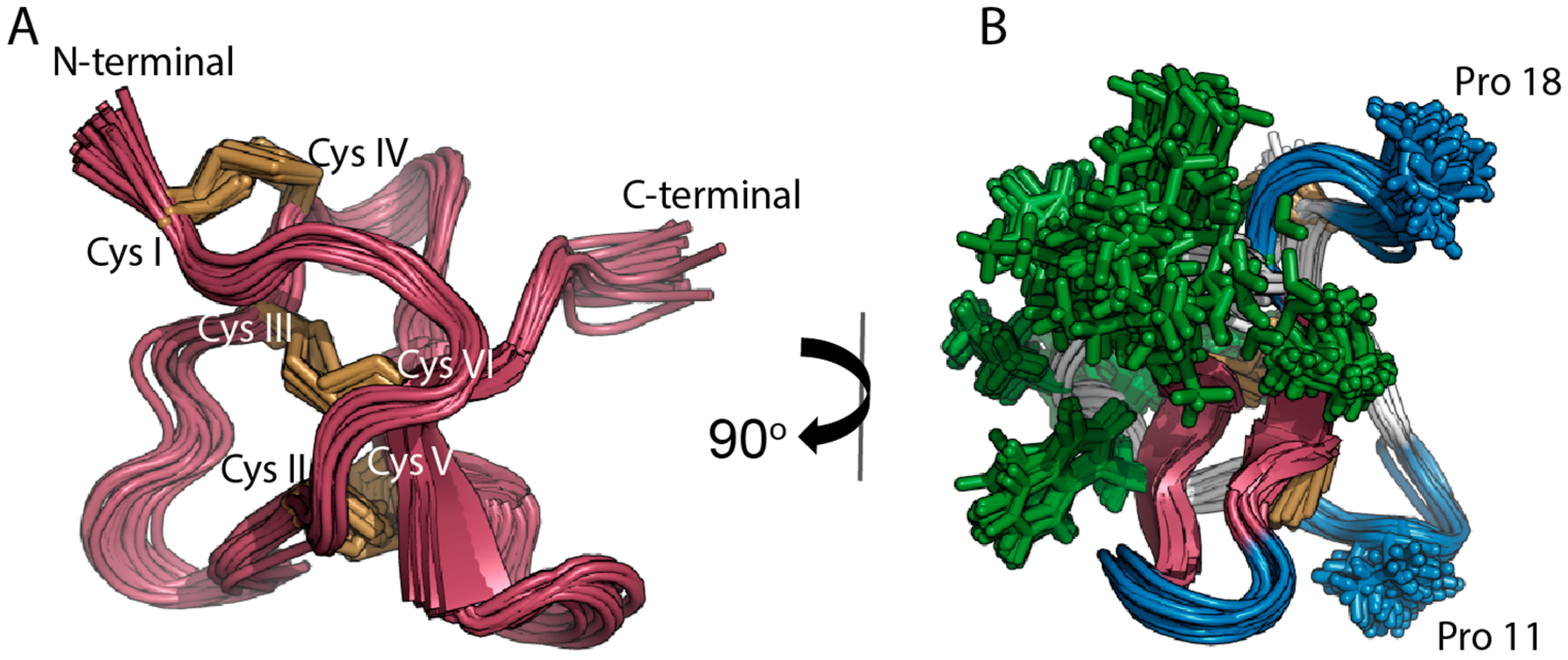
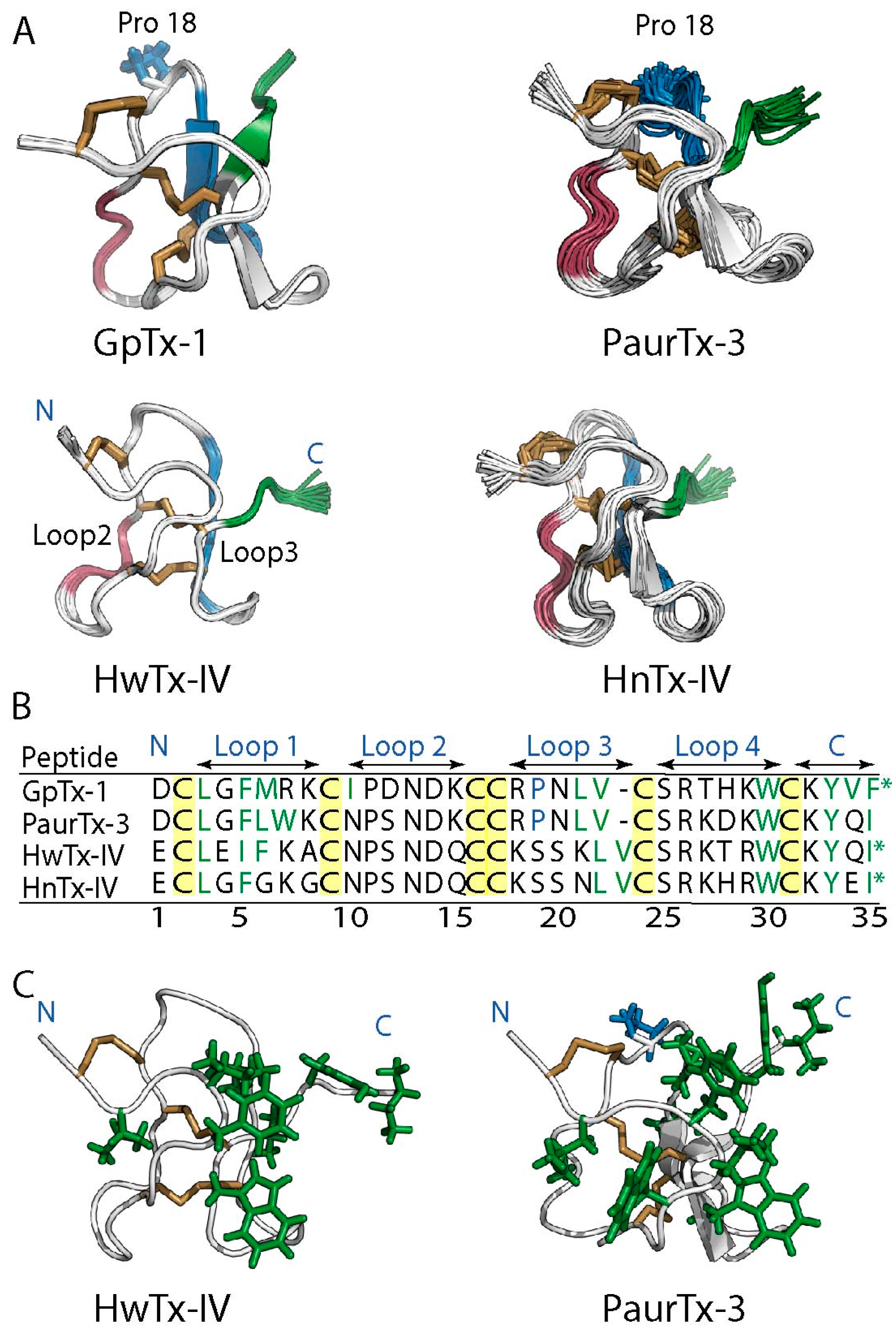
2.1. Solution NMR Structure of PaurTx-3
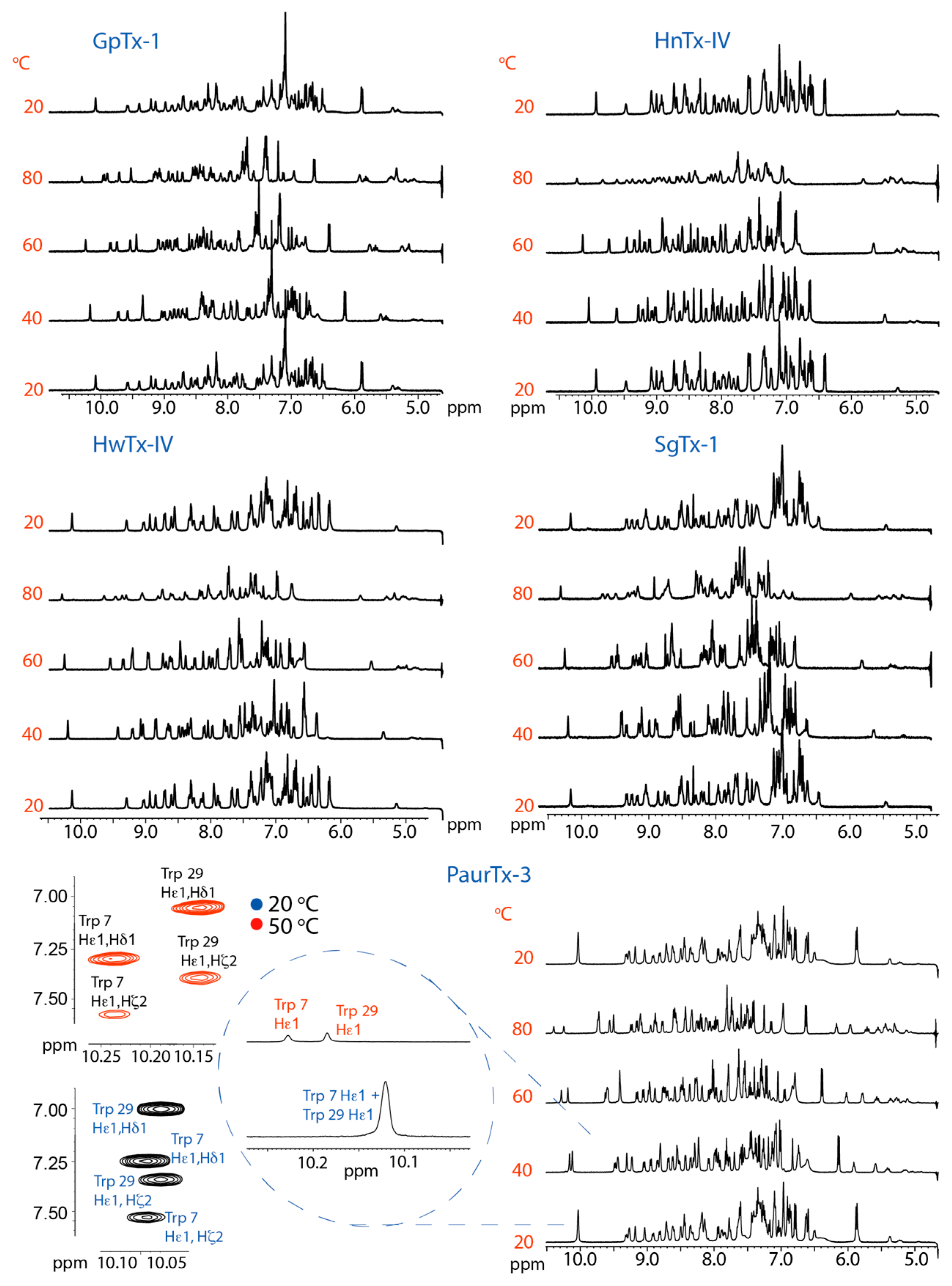
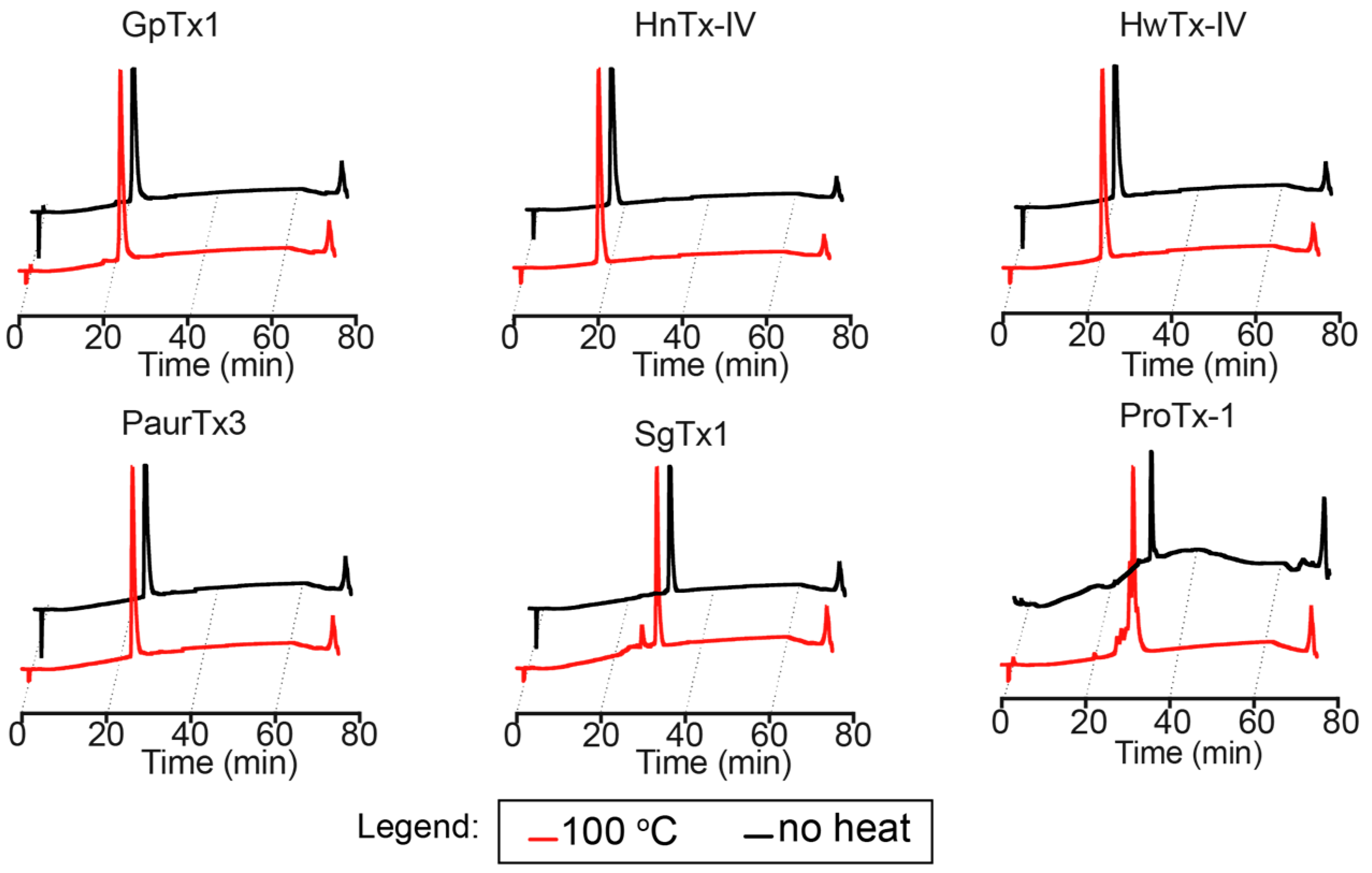
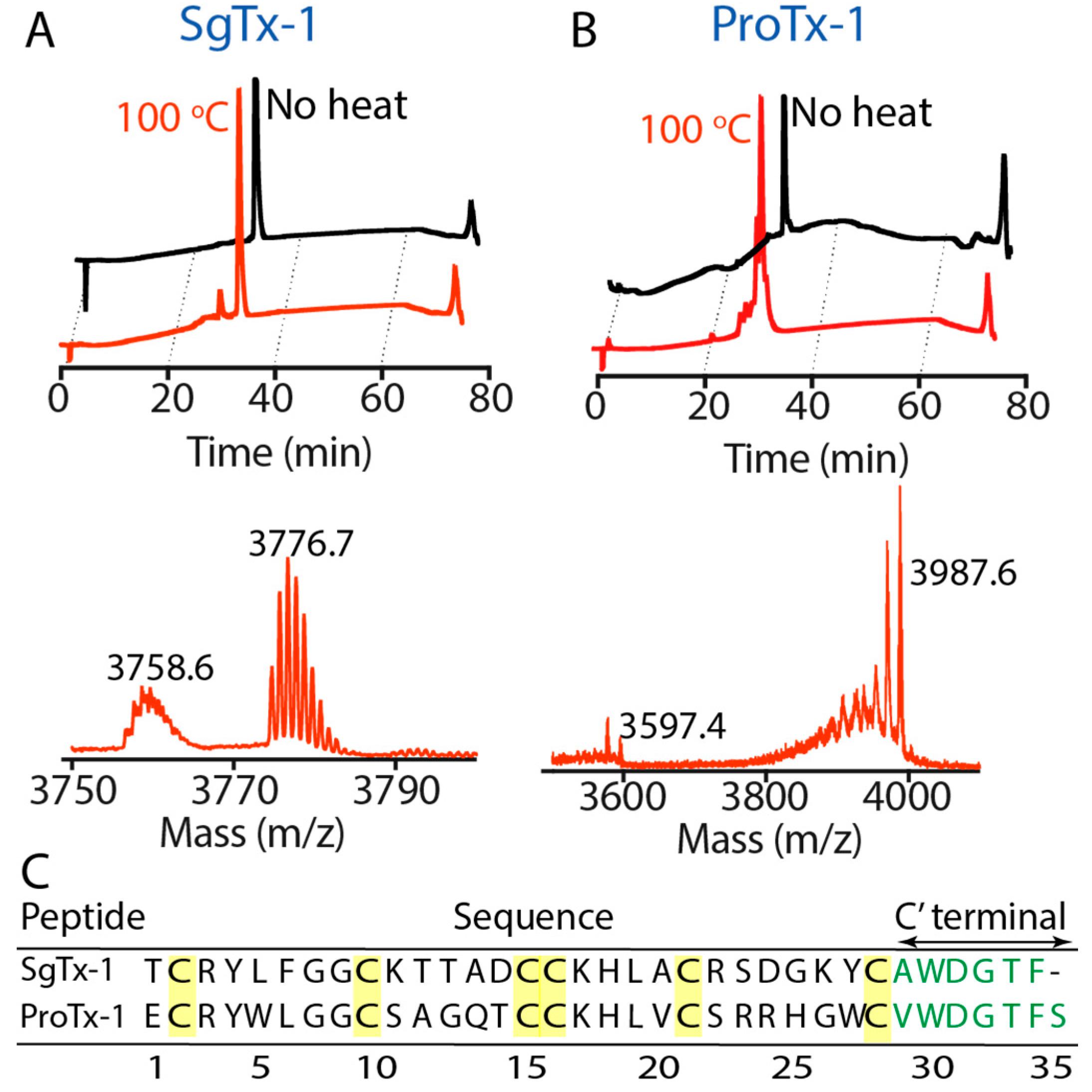
2.2. Thermal Stability
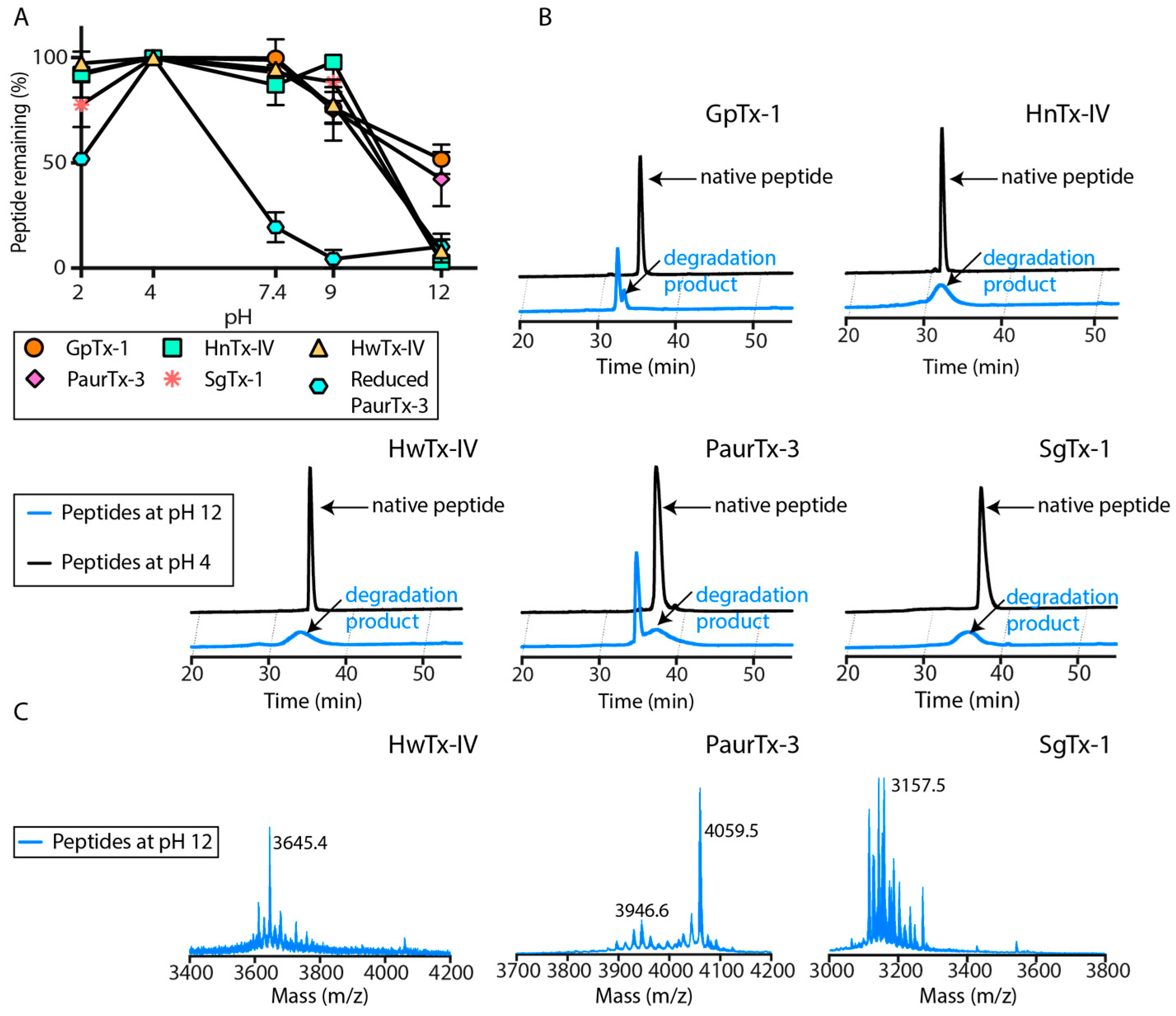
2.3. pH Dependent Hydrolysis
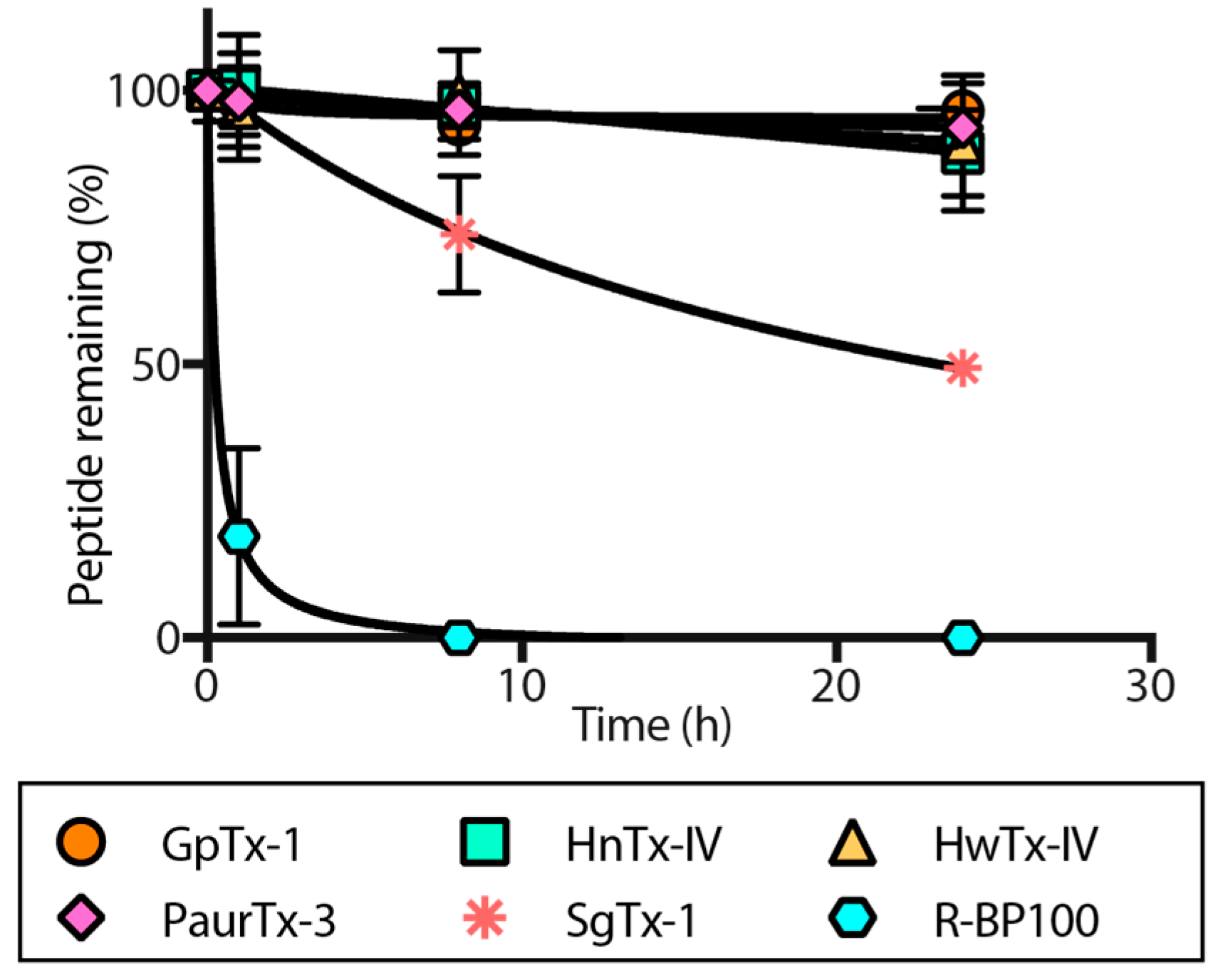
2.4. Proteolytic Degradation
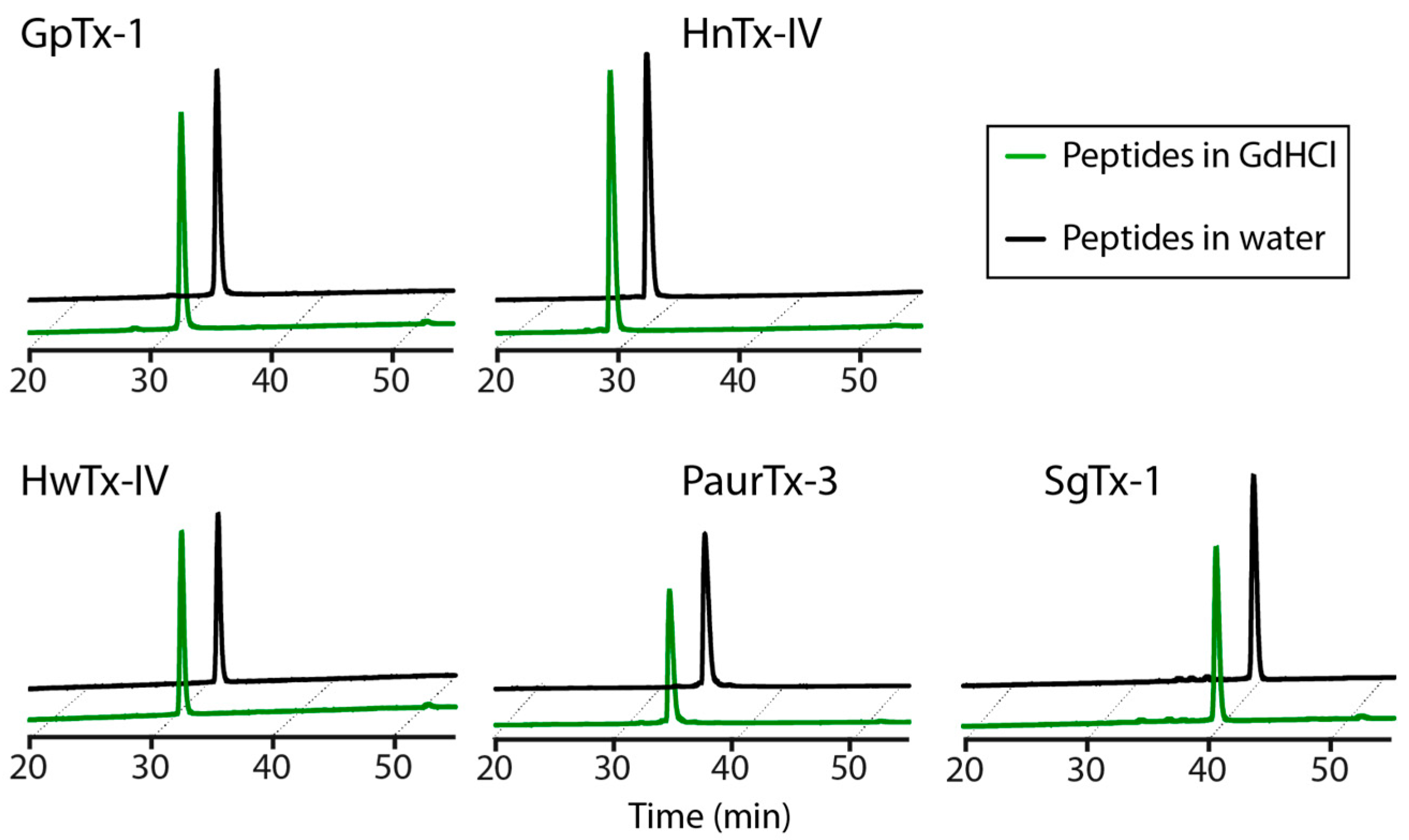
2.5. Chaotropic Degradation
2.6. Root-Mean-Square Deviation at the C-Termini GMTs
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Peptide Synthesis
5.2. NMR Structure Calculation for PaurTx-3
5.3. Peptide Quantification
5.4. Analytical RP-HPLC
5.5. Thermal Stability
5.6. pH Dependent Hydrolysis
5.7. Proteolytic Degradation
5.8. Chaotropic Stability
5.9. RMSD Calculation
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smith, J.J.; Lau, C.H.Y.; Herzig, V.; Ikonomopoulou, M.P.; Rash, L.D.; King, G.F. Therapeutic applications of spider-venom peptides. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; King, G.F., Ed.; Royal Society of Chemistry: London, UK, 2015; pp. 221–244. [Google Scholar]
- Escoubas, P.; Bosmans, F. Spider peptide toxins as leads for drug development. Expert Opin. Drug Discov. 2007, 2, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Cestele, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetter, I.; Deuis, J.R.; Mueller, A.; Israel, M.R.; Starobova, H.; Zhang, A.; Rash, L.D.; Mobli, M. NaV1.7 as a pain target—From gene to pharmacology. Pharmacol. Ther. 2017, 172, 73–100. [Google Scholar] [CrossRef] [PubMed]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the NaV1.1 channel in mechanical pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Vetter, I.; Cardoso, F.C.; Inserra, M.; King, G. Does nature do ion channel drug discovery better than us? In Ion Channel Drug Discovery; Cox, B., Gosling, M., Eds.; Royal Society of Chemistry: London, UK, 2015; pp. 297–319. [Google Scholar]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Flinspach, M.; Xu, Q.; Piekarz, A.D.; Fellows, R.; Hagan, R.; Gibbs, A.; Liu, Y.; Neff, R.A.; Freedman, J.; Eckert, W.A.; et al. Insensitivity to pain induced by a potent selective closed-state NaV1.7 inhibitor. Sci. Rep. 2017, 7, 39662. [Google Scholar] [CrossRef] [PubMed]
- Netirojjanakul, C.; Miranda, L.P. Progress and challenges in the optimization of toxin peptides for development as pain therapeutics. Curr. Opin. Chem. Biol. 2017, 38, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Osteen, J.D.; Sampson, K.; Iyer, V.; Julius, D.; Bosmans, F. Pharmacology of the Nav1.1 domain IV voltage sensor reveals coupling between inactivation gating processes. Proc. Natl. Acad. Sci. USA 2017, 114, 6836–6841. [Google Scholar] [PubMed]
- Agwa, A.J.; Henriques, S.T.; Schroeder, C.I. Gating modifier toxin interactions with ion channels and lipid bilayers: is the trimolecular complex real? Neuropharmacology 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Tedford, H.W.; Maggio, F. Structure and function of insecticidal neurotoxins from Australian funnel-web spiders. J. Toxicol. Toxin Rev. 2002, 21, 361–389. [Google Scholar] [CrossRef]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar] [CrossRef]
- Pallaghy, P.K.; Nielsen, K.J.; Craik, D.J.; Norton, R.S. A common structural motif incorporating a cystine knot and a triple-stranded beta-sheet in toxic and inhibitory polypeptides. Protein Sci. 1994, 3, 1833–1839. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.K.; Ligutti, J.; Liu, D.; Zou, A.; Poppe, L.; Li, H.; Andrews, K.L.; Moyer, B.D.; McDonough, S.I.; Favreau, P.; et al. Engineering potent and selective analogues of GpTx-1, a tarantula venom peptide antagonist of the NaV1.7 sodium channel. J. Med. Chem. 2015, 58, 2299–2314. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Kim, S.; Roh, S.H.; Endoh, H.; Kodera, Y.; Maeda, T.; Kohno, T.; Wang, J.M.; Swartz, K.J.; Kim, J.I. Solution structure and functional characterization of SGTx1, a modifier of KV2.1 channel gating. Biochemistry 2004, 43, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xiao, Y.; Xu, X.; Xiong, X.; Lu, S.; Liu, Z.; Zhu, Q.; Wang, M.; Gu, X.; Liang, S. Structure-activity relationships of hainantoxin-IV and structure determination of active and inactive sodium channel blockers. J. Biol. Chem. 2004, 279, 37734–37740. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.T.; Deplazes, E.; Lawrence, N.; Cheneval, O.; Chaousis, S.; Inserra, M.; Thongyoo, P.; King, G.F.; Mark, A.E.; Vetter, I.; et al. Interaction of tarantula venom peptide ProTx-II with lipid membranes is a prerequisite for its inhibition of human voltage-gated sodium channel NaV1.7. J. Biol. Chem. 2016, 29, 17049–17065. [Google Scholar] [CrossRef] [PubMed]
- Minassian, N.A.; Gibbs, A.; Shih, A.Y.; Liu, Y.; Neff, R.A.; Sutton, S.W.; Mirzadegan, T.; Connor, J.; Fellows, R.; Husovsky, M.; et al. Analysis of the structural and molecular basis of voltage-sensitive sodium channel inhibition by the spider toxin huwentoxin-IV (mu-TRTX-Hh2a). J. Biol. Chem. 2013, 288, 22707–22720. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Dekan, Z.; Wingerd, J.S.; Smith, J.J.; Munasinghe, N.R.; Bhola, R.F.; Imlach, W.L.; Herzig, V.; Armstrong, D.A.; Rosengren, K.J.; et al. Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide Pn3a. Sci. Rep. 2017, 7, 40883. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Smith, J.J.; Vetter, I.; Rupasinghe, D.B.; Er, S.Y.; Senff, S.; Herzig, V.; Mobli, M.; Lewis, R.J.; Bosmans, F.; et al. Seven novel modulators of the analgesic target NaV1.7 uncovered using a high-throughput venom-based discovery approach. Br. J. Pharmacol. 2015, 172, 2445–2458. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Rash, L.; Zhu, S.; Diochot, S.; Lazdunski, M.; Escoubas, P.; Tytgat, J. Four novel tarantula toxins as selective modulators of voltage-gated sodium channel subtypes. Mol. Pharmacol. 2006, 69, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Mandard, N.; Bulet, P.; Caille, A.; Daffre, S.; Vovelle, F. The solution structure of gomesin, an antimicrobial cysteine-rich peptide from the spider. Eur. J. Biochem. 2002, 269, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Agwa, A.J.; Lawrence, N.; Deplazes, E.; Cheneval, O.; Chen, R.; Craik, D.J.; Schroeder, C.I.; Henriques, S.T. Spider peptide toxin HwTx-IV engineered to bind to lipid membranes has an increased inhibitory potency at human voltage-gated sodium channel hNaV1.7. Biochim. Biophys. Acta 2017, 1859, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Deplazes, E.; Henriques, S.T.; Smith, J.J.; King, G.F.; Craik, D.J.; Mark, A.E.; Schroeder, C.I. Membrane-binding properties of gating modifier and pore-blocking toxins: Membrane interaction is not a prerequisite for modification of channel gating. Biochim. Biophys. Acta 2016, 1858, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.Y.; King, G.F.; Mobli, M. Molecular basis of the interaction between gating modifier spider toxins and the voltage sensor of voltage-gated ion channels. Sci. Rep. 2016, 6, 34333. [Google Scholar] [CrossRef] [PubMed]
- Revell Phillips, L.; Milescu, M.; Li-Smerin, Y.; Mindell, J.A.; Kim, J.I.; Swartz, K.J. Voltage-sensor activation with a tarantula toxin as cargo. Nature 2005, 436, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; MacKinnon, R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 2004, 430, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Salari, A.; Vega, B.S.; Milescu, L.S.; Milescu, M. Molecular interactions between tarantula toxins and low-voltage-activated calcium channels. Sci. Rep. 2016, 6, 23894. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; King, G.F. The cystine knot is responsible for the exceptional stability of the insecticidal spider toxin omega-hexatoxin-Hv1a. Toxins 2015, 7, 4366–4380. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Martin-Eauclaire, M.-F.; Swartz, K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 2008, 456, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Swartz, K.J. Targeting sodium channel voltage sensors with spider toxins. Trends Pharmacol. Sci. 2010, 31, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., 3rd; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–383. [Google Scholar] [CrossRef] [PubMed]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55, 29–32. [Google Scholar] [CrossRef]
- Torcato, I.M.; Huang, Y.H.; Franquelim, H.G.; Gaspar, D.; Craik, D.J.; Castanho, M.A.; Troeira Henriques, S. Design and characterization of novel antimicrobial peptides, R-BP100 and RW-BP100, with activity against Gram-negative and Gram-positive bacteria. Biochim. Biophys. Acta 2013, 1828, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Colgrave, M.L.; Craik, D.J. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef] [PubMed]
- Er, S.Y.; Cristofori-Armstrong, B.; Escoubas, P.; Rash, L.D. Discovery and molecular interaction studies of a highly stable, tarantula peptide modulator of acid-sensing ion channel 1. Neuropharmacology 2017. [Google Scholar] [CrossRef] [PubMed]
- Hardy, M.C.; Daly, N.L.; Mobli, M.; Morales, R.A.V.; King, G.F. Isolation of an orally active insecticidal toxin from the venom of an australian tarantula. PLoS ONE 2013, 8, e73136. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Scholtz, J.M. Guanidine hydrochloride unfolding of peptide helices: separation of denaturant and salt effects. Biochemistry 1996, 35, 7292–7297. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.E.; Dempsey, C.E.; Neilson, G.W.; Kline, S.R.; Brady, J.W. Preferential interactions of guanidinum ions with aromatic groups over aliphatic groups. J. Am. Chem. Soc. 2009, 131, 16689–16696. [Google Scholar] [CrossRef] [PubMed]
- Cobos, E.S.; Filimonov, V.V.; Gálvez, A.; Valdivia, E.; Maqueda, M.; Martı́nez, J.C.; Mateo, P.L. The denaturation of circular enterocin AS-48 by urea and guanidinium hydrochloride. Biochim. Biophys. Acta 2002, 1598, 98–107. [Google Scholar] [CrossRef]
- Liu, Y.; Li, D.; Wu, Z.; Li, J.; Nie, D.; Xiang, Y.; Liu, Z. A positively charged surface patch is important for hainantoxin-IV binding to voltage-gated sodium channels. J. Pept. Sci. 2012, 18, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Bingham, J.P.; Zhu, W.; Moczydlowski, E.; Liang, S.; Cummins, T.R. Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain II voltage sensor in the closed configuration. J. Biol. Chem. 2008, 283, 27300–27313. [Google Scholar] [CrossRef] [PubMed]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley Interscience: New York, NY, USA, 1986. [Google Scholar]
- Güntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar] [PubMed]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Nederveen, A.J.; Doreleijers, J.F.; Vranken, W.; Miller, Z.; Spronk, C.A.; Nabuurs, S.B.; Güntert, P.; Livny, M.; Markley, J.L.; Nilges, M.; et al. RECOORD: A recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins 2005, 59, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Brünger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystall. D Biol. Crystall. 1998, 54, 905–921. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energies (kcal/mol) | |
| Overall | −1232.31 ± 50.68 |
| Bonds | 23.83 ± 2.03 |
| Angles | 64.83 ± 6.52 |
| Improper | 19.89 ± 2.47 |
| Dihedral | 164.27 ± 1.66 |
| Van der Waals | −131.51 ± 7.15 |
| Electrostatic | −1374.68 ± 52.43 |
| NOE | 0.38 ± 0.04 |
| Constrained dihedral (cDih) | 0.66 ± 0.40 |
| MolProbity Statistics | |
| Clash score (>0.4 Å/1000 atoms) | 11.10 ± 3.94 |
| Poor rotamers (%) | 2.88 ± 2.30 |
| Ramachandran outliers (%) | 0.47 ± 1.14 |
| Ramachandran favoured (%) | 87.50 ± 3.36 |
| MolProbity score | 2.45 ± 0.26 |
| MolProbity percentile 3 | 50.40 ± 14.91 |
| Atomic RMSD (Å) | |
| Mean global backbone (2–31) 4 | 0.71 ± 0.21 |
| Mean global heavy (2–31) | 1.51 ± 0.21 |
| Mean global backbone (1–34) | 0.88 ± 0.23 |
| Mean global heavy (1–34) | 1.67 ± 0.20 |
| Distance Restraints | |
| Intraresidue (i − j = 0) | 134 |
| Sequential (|i − j| = 1) | 120 |
| Medium range (|i − j| < 5) | 42 |
| Long range (|i − j| > 5) | 40 |
| Hydrogen bonds 5 | 14 |
| Total | 350 |
| Dihedral Angle Restraints | |
| φ | 18 |
| ψ | 18 |
| χ1 | 5 |
| Total | 41 |
| Violations from Experimental Restraints | |
| Total NOE violations exceeding 0.2 Å | 1 |
| Total dihedral violations exceeding 2.0° | 2 |
| Peptide | Backbone RMSD (Å) (Pre C-Term) 2 | Backbone RMSD (Å) (C-Term) 3 |
|---|---|---|
| GpTx-1 | 0.18 ± 0.09 (Asp 1–Trp 29) | 0.17 ± 0.09 (Lys 31–Phe 34) |
| HnTx-IV | 0.60 ± 0.13 (Glu 1–Trp 30) | 0.51 ± 0.22 (Lys 32–Ile 35) |
| HwTx-IV | 0.34 ± 0.09 (Glu 1–Trp 30) | 0.39 ± 0.17 (Lys 32–Ile 35) |
| PaurTx-3 | 0.79 ± 0.22 (Asp 1–Trp 29) | 0.30 ± 0.14 (Lys 31–Ile 34) |
| SgTx-1 | 0.38 ± 0.11 (Thr 1–Tyr 27) | 1.58 ± 0.43 (Asp 29–Phe 34) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agwa, A.J.; Huang, Y.-H.; Craik, D.J.; Henriques, S.T.; Schroeder, C.I. Lengths of the C-Terminus and Interconnecting Loops Impact Stability of Spider-Derived Gating Modifier Toxins. Toxins 2017, 9, 248. https://doi.org/10.3390/toxins9080248
Agwa AJ, Huang Y-H, Craik DJ, Henriques ST, Schroeder CI. Lengths of the C-Terminus and Interconnecting Loops Impact Stability of Spider-Derived Gating Modifier Toxins. Toxins. 2017; 9(8):248. https://doi.org/10.3390/toxins9080248
Chicago/Turabian StyleAgwa, Akello J., Yen-Hua Huang, David J. Craik, Sónia T. Henriques, and Christina I. Schroeder. 2017. "Lengths of the C-Terminus and Interconnecting Loops Impact Stability of Spider-Derived Gating Modifier Toxins" Toxins 9, no. 8: 248. https://doi.org/10.3390/toxins9080248
APA StyleAgwa, A. J., Huang, Y. -H., Craik, D. J., Henriques, S. T., & Schroeder, C. I. (2017). Lengths of the C-Terminus and Interconnecting Loops Impact Stability of Spider-Derived Gating Modifier Toxins. Toxins, 9(8), 248. https://doi.org/10.3390/toxins9080248