1. Introduction
The detection of volatile organic compounds (VOCs) has application in a wide range of industries including food, agriculture, health and security. While conventional detection technologies such as semiconductor based solid state e-nose systems [
1,
2] are progressing, there is growing interest in employing chemosensory protein-based biosensors available in olfactory systems in nature as the recognition elements with a view to exploit their superior detection sensitivity and selectivity [
3,
4,
5,
6]. Towards the ultimate goal of developing next-generation VOC detection systems, the use of whole-cell assays [
7], odorant binding proteins [
8], olfactory receptors [
9,
10], peptides [
11] and aptamers [
12] have been proposed. One of the key challenges in using these biosensors in VOC-detection systems is the requirement for an efficient gas sampling interface to transfer the VOCs from the gas phase into the liquid phase. This is because the biosensors are made of proteins which are functional only in aqueous solutions. This problem is further complicated by the physicochemical properties of certain volatile compounds, which are of significant interest in the health and security domains. The properties, such as lipophilicity and low water solubility, limit the transfer efficiency of the compounds into an aqueous solution, therefore reducing the overall sensitivity of the VOC detection system. To use biosensors as part of field-deployable the VOC detection systems, a well-characterised gas-to-liquid transfer strategy needs to be established and the key factors influencing VOC capture efficiency need to be identified to enable system optimization.
A number of studies examining gas-to-liquid transfer methods for the detection of the VOCs by biosensors have been published. Park et al. [
13] used a porous polycarbonate membrane separating a gas flow chamber and olfactory cells to develop a platform for mimicking the human olfactory system. Stemme et al. [
14] used a microfabricated perforated silicon membrane to establish a gas-liquid interface for subsequent detection using antibodies. Toda et al. [
15] used a polydimethyl siloxane (PDMS) membrane to construct a micro scrubber. However, mass transfer through membranes is a limiting factor for detection speed and collection efficiency [
16]. Furthermore, there is a risk of adsorption of the VOCs on the membrane material, causing contamination and sample carry over.
Miniature systems based on open channels that do not rely on any membrane structure have also been reported [
17,
18]. These systems provide an effective way to establish a large gas-liquid interface area and eliminate issues related with membranes. These interfaces used surface wettability to spread a small volume of liquid over an area. However, in these studies, maintaining the gas-liquid interface required complex pumping systems for interface stabilization which prevents the interface from drying and the liquid phase from flooding over the open channel structure. The requirement for pumping systems increases the overall complexity and makes it difficult to deploy the technology for industrial applications. There is a desire to develop simpler liquid-handling strategies for utilising gas-liquid interfaces in open channels. The volatile capture efficiency of these interfaces has never been systematically studied, especially for the case of lipophilic molecules with low water solubility.
This study reports a gas-sampling interface based on gas-to-liquid extraction using an open capillary channel device and this study further demonstrates capillary flow-driven loading and collection of capture liquid without using pumps. The pumpless operation provides simplicity by eliminating the requirement for any external active pressure control system, making it more desirable for field deployable applications. By using this device as a model system, the key factors influencing VOC extraction efficiency for lipophilic VOCs were identified. The impact of parameters such as sampling temperature, gas flow rate and chemical modification of the capture liquid composition have been studied.
1.1. Device Structure and Operation
A schematic of our microfabricated, open-channel device is shown in
Figure 1a, with an image of a test device shown in
Figure 1b. The device has external dimensions of 65 × 30 × 3.3 mm and is composed of three layers of bonded borosilicate glass. The device includes an inlet and outlet for the liquid phase and two circular ports (4 mm diameter) for introducing the gas sample. The device is designed such that the gas sample can be passed through the device across the liquid surface in the open-channel network at a controlled flow rate. The volume of the gas chamber is approximately 500 µL and the open channel network holds 30 µL of capture liquid. The gas-liquid interface area is approximately 3 cm
2. For the initial priming, the liquid is introduced into the device through the liquid inlet on the left side. The liquid spreads throughout the open channel network by capillary action. The details of the open channel network design are shown in
Figure 2a. The design involves hexagonal protrusions between which the capture liquid sits.
Figure 2b shows the dimensions of the repeating unit cell. A scanning electron microscope (SEM) image of the capillary bed is shown in
Figure 2c. An optical microscope view of the device is shown in
Figure 2d. Gas sampling is achieved by introducing the gas sample through the device via the gas ports. During this process, the VOCs in the gas phase are transferred into the liquid in the open channel. The capture liquid is collected from the device for analysis via the outlet with a gas tight syringe (see
Section 2.6 for details of collection procedure).
1.2. Computational Modelling
The use of capillary flow significantly simplifies device operation by eliminating the requirement for an external pumping system. Capillary flow depends on the surface wettability dictated by contact angle. Computational modelling was used to analyse the contact angle dependence of the capillary flow. COMSOL Multiphysics (
www.comsol.com) with the level set method was used to model two-phase flow and track movement of the air-water interface. To minimize computation time, a simplified 2D analysis of the repeating unit cell illustrated in
Figure 2b was used as the model geometry.
Figure 3 shows the details of the model geometry. The inlet and outlet boundary conditions were set to zero pressure for spontaneous flow condition. The symmetry boundary condition was selected for the top and bottom walls, effectively modelling an infinite 2D expansion of the repeating pattern. The wetted wall boundary conditions in the Multiphysics interface provide user set contact angles. The contact angle range between 5 and 60 degrees was studied.
Using the fluid dynamics interface in COMSOL, the model is solved with two sets of governing equations. The level set method uses Equation (1) to solve the moving interface, where φ is the level set function which is equal to zero in one fluid domain (water) and equal to one in the other (air). Across the interface, there is a continuous transition from zero to one. The velocity field of the interface is represented by u. The parameter ε determines the thickness of the region where φ goes smoothly from zero to one and is set to the same order as the size of the mesh elements. γ is the re-initialization parameter, which is set to the maximum magnitude of the velocity field u.
The other set of equations solved in the model are continuity and Navier-Stokes Equations (2) and (3), which describe the mass and momentum transport for fluids. The surface tension is included in the model to account for capillary effects. ρ denotes the density, μ equals the dynamic viscosity, u represents velocity, and p denotes pressure. Fst is the surface tension acting at the air-water interface.
2. Materials and Methods
2.1. Fabrication
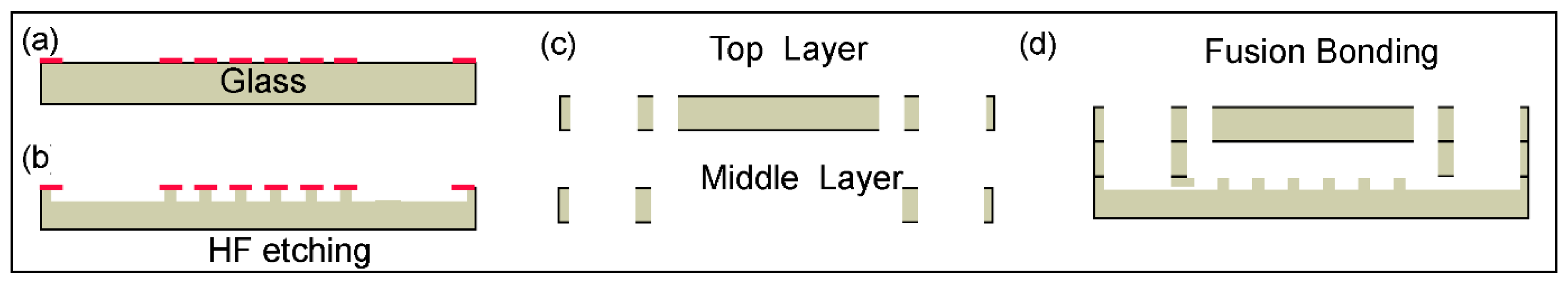
The 3-layer device was fabricated from borosilicate glass (1.1 mm thickness) using standard microfabrication techniques. First, a patterned Cr/Au metal layer was fabricated on the glass substrate using photolithography and etching (
Figure 4a). The process was followed by hydrofluoric acid (HF), etching to a depth of 100 µm for creating the structures that form the open channel network (
Figure 4b). The middle layer and the top layers were fabricated using laser machining (
Figure 4c). Finally, the three-layer stack was fusion bonded by annealing at 700 °C (
Figure 4d).
2.2. Surface Preparation and Maintenance
As the device is based on capillary flow, stable operation relies on maintaining the water wettability of the open channel surface. The wettability of the borosilicate glass surface was achieved by cleaning the surface in aqueous NaOH solution (100 mg·mL−1) to generate OH groups on the glass surface. This cleaning procedure was used whenever surface wettability needed to be restored. The fabricated device was immersed in the solution overnight while making sure the surface of the open channel was completely covered by solution without residual bubbles. The device was rinsed with Milli-Q water to wash out remaining NaOH. Surface wettability was confirmed by priming the device with food colouring solution and observing liquid spreading in the open-channel network. For long term storage, the surface was wetted with Milli-Q water and all the ports on the device were sealed with Kapton tape to prevent evaporation and surface drying.
2.3. Model Volatiles
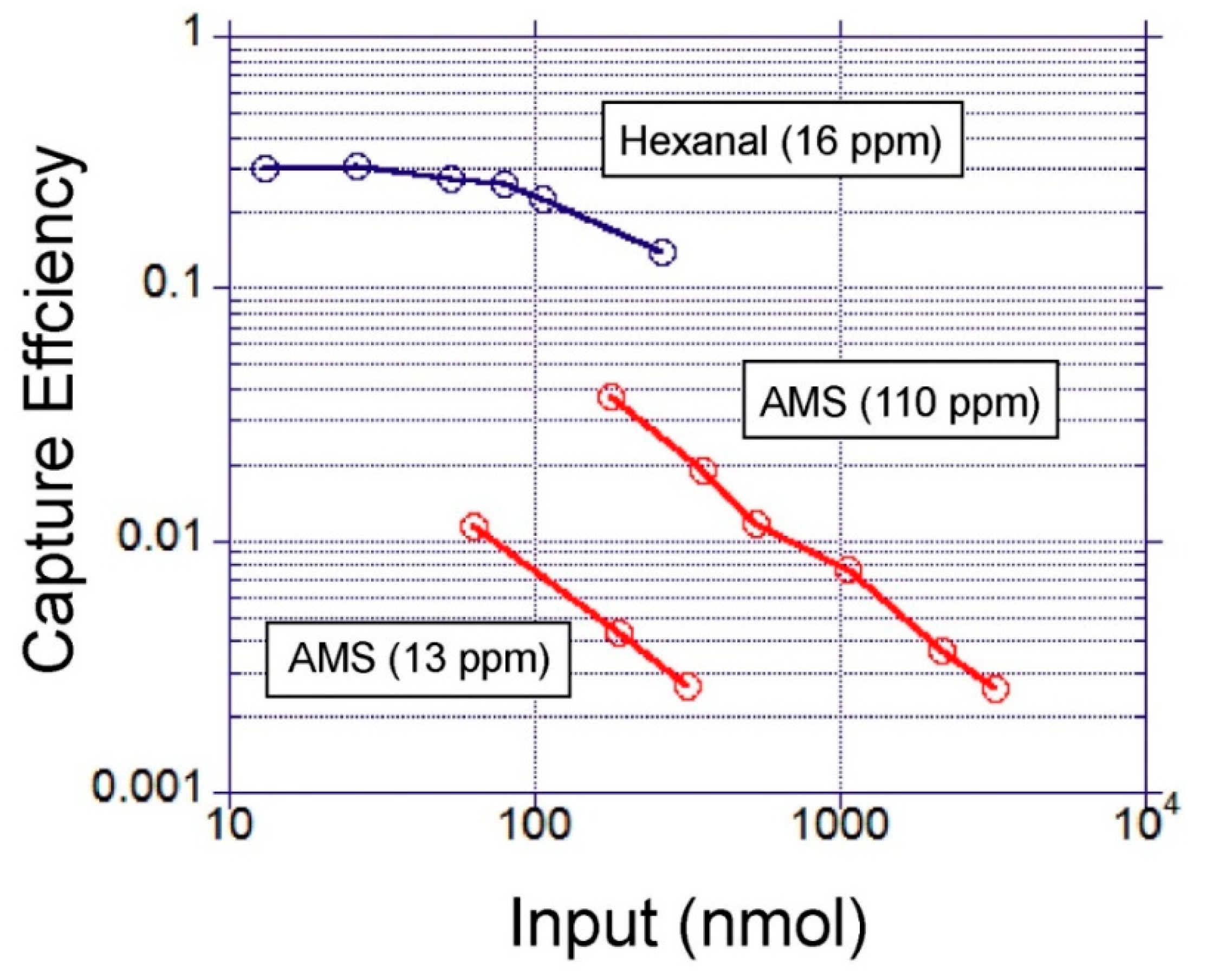
The characterization of gas-to-liquid extraction efficiency was performed using gas samples spiked with model volatiles.
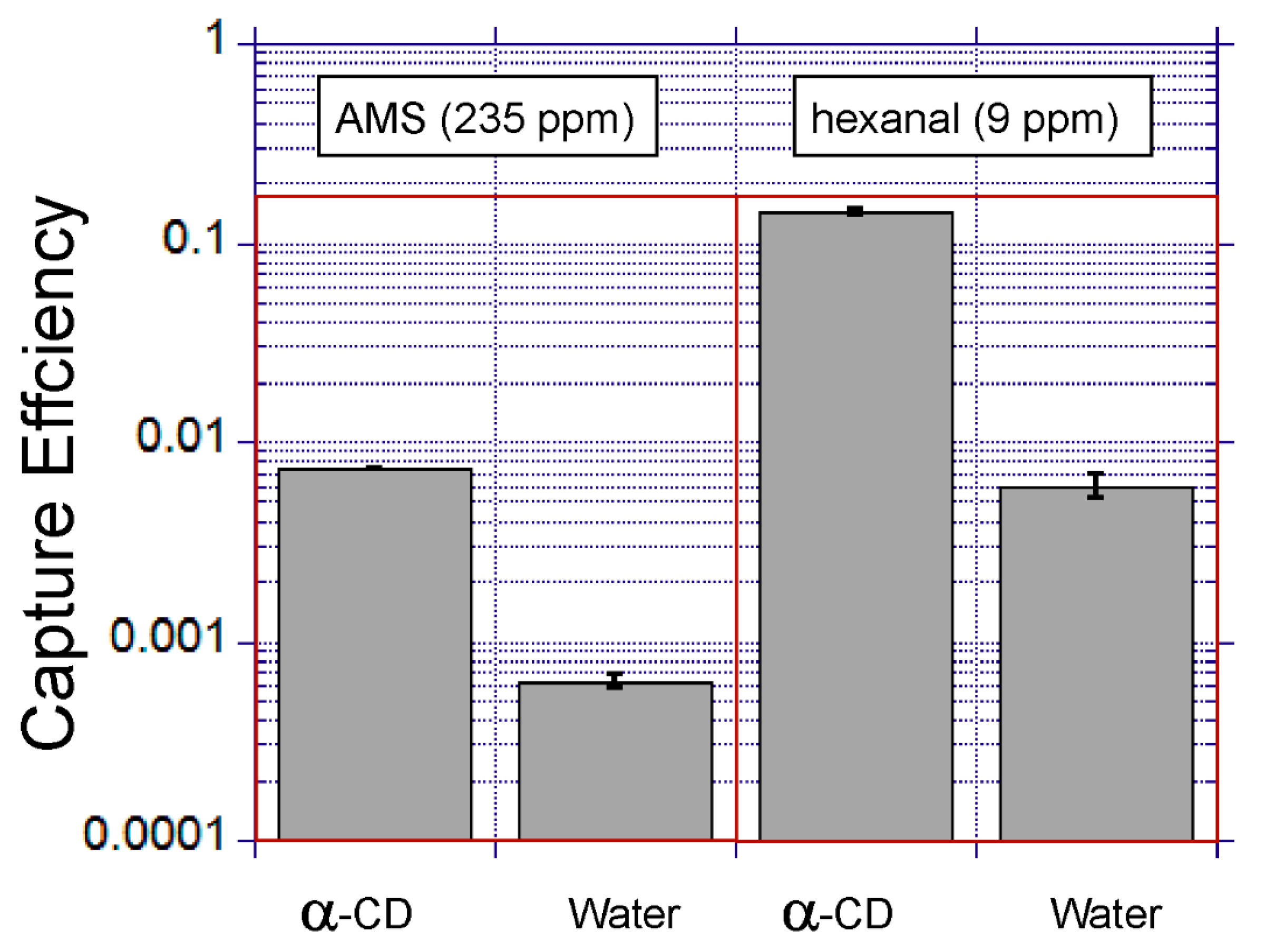
Table 1 shows the selected volatiles and their properties. The two model volatiles with distinct physicochemical properties were selected to assess the impact of compound lipophilicity on the gas-liquid extraction efficiency. Hexanal and allyl methyl sulfide are also known as breath biomarkers for lung cancer and malaria, respectively.
2.4. Gas Sample Preparation
A 50 mL round bottomed glass flask with an outlet and inlet port was heated to 200 °C in a bead bath on a hotplate, well above the boiling points of the volatile compounds. The VOC (3 µL) was injected into the sealed flask through the inlet port to vaporize the compound. The flask was allowed to equilibrate at this temperature for 5 min, after which nitrogen gas was passed through the flask inlet port at a rate of 50 mL min−1. The nitrogen line was equipped with a gas flow meter (TSI Inc., Shoreview, MN, USA, SKU: 4146) with a volumetric flow rate display to monitor the total volume of the gas added. The gas sample bag (SKC Inc., Eighty Four, PA, USA, SamplePro FlexFilm sample bag with volumes: 1/3/5 L) was connected to the flask outlet to collect the VOC spiked gas sample. The gas flow was stopped when the capacity of the sample bag was reached. For further dilutions, 100 mL of the initial bag contents was injected into a separate bag and nitrogen was added depending on the dilution level needed. The freshly prepared gas sample bags containing nitrogen and the volatile compound were used for the experiments. The concentrations of the volatiles in the bag were confirmed by sampling 10 mL of gas in a sealed GC vial and analysing with a GC-MS (Agilent 6890 gas chromatograph (GC) coupled with an Agilent 5973 mass selective (MS) detector and automatic headspace sampler (7694E)).
2.5. Capture Liquid Solution
The capture solution was 100 mg mL−1 alpha-cyclodextrin (Sigma, Aldrich, MI, USA, C4642) in pure Milli-Q water, unless otherwise indicated. The solution was freshly prepared each day.
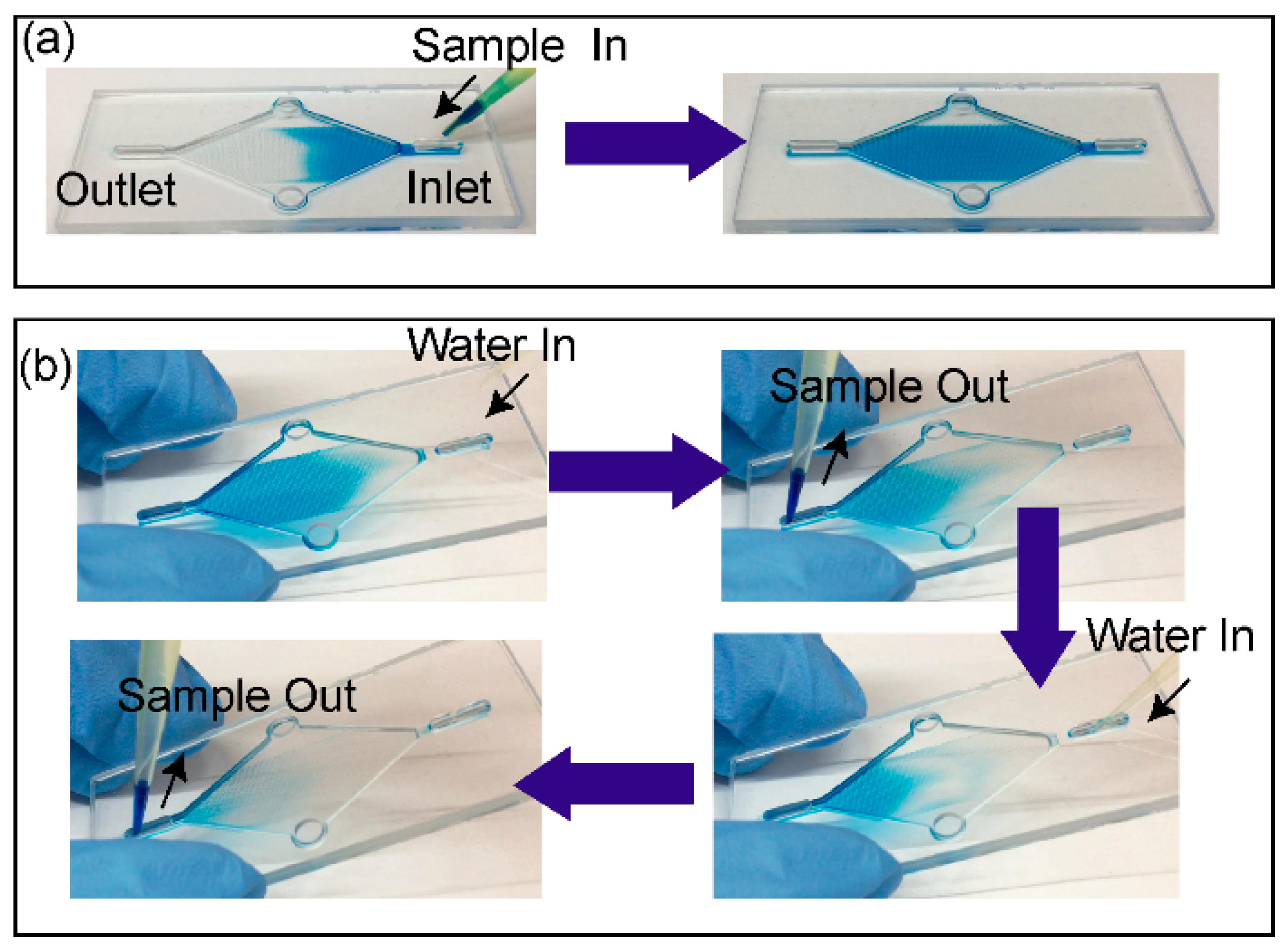
2.6. Device Priming and Capture Liquid Collection
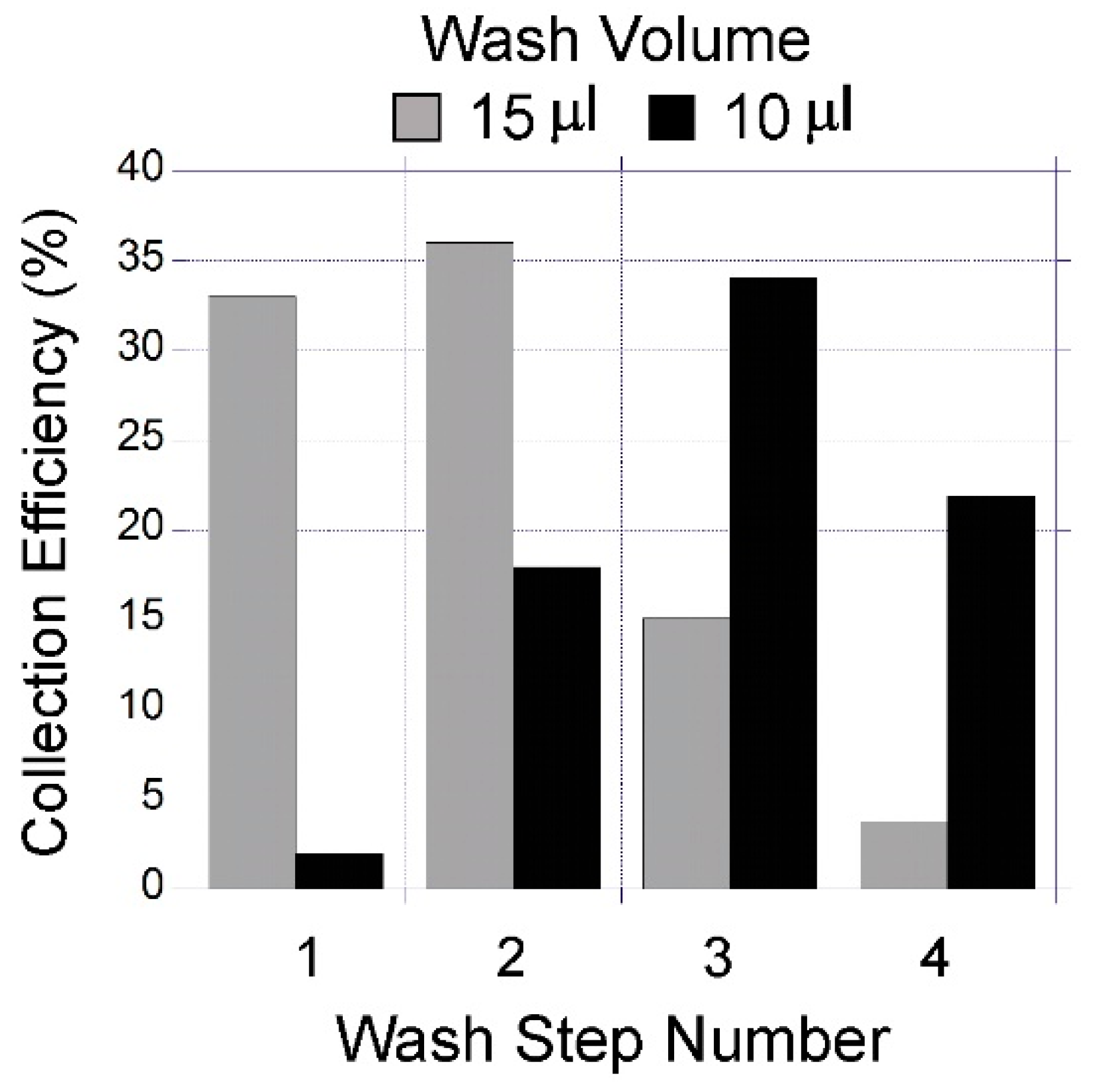
The spontaneous capillary flow was used to prime the chip, and for loading and collection of the capture liquid. The open channel was initially primed with water, then 15 µL of capture liquid was introduced through the inlet after which the water at the outlet was removed by pipette. This procedure was repeated by introducing an additional 15 µL of capture liquid to complete the loading. Once the loading was complete, the inlet and outlet ports were emptied completely by pipette. At this stage, the gas-liquid interface was stabilised by surface tension and the device was ready for gas sampling. To collect a 30 µL capture solution following exposure to the VOC, it was displaced by introducing 2 × 15 µL water at the inlet. This corresponds to approximately 30 µL of the capture liquid sample collected from the device for each measurement. (
Supplementary video is available demonstrating capture liquid loading and collection.)
2.7. Gas Sampling
The device was loaded with fresh capture liquid. The inlet and outlet ports for liquid were sealed with Kapton tape. The gas sampling was performed as illustrated in
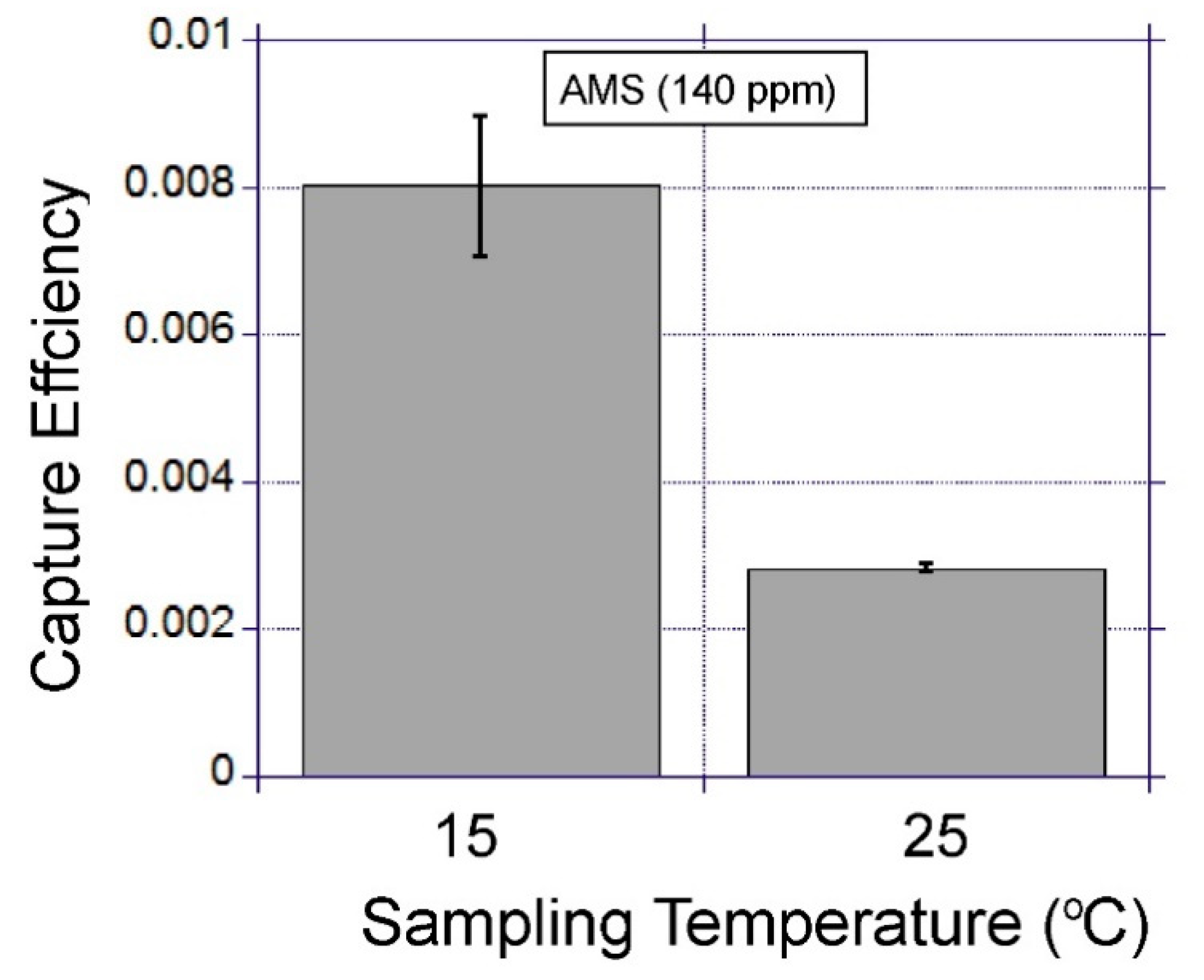
Figure 5. The device was placed on a copper plate attached to a thermo-electric cooler with a control unit (PLC-24V6A, Kurag Electronics, Tokyo, Japan) which allowed temperature adjustment between 15 °C and 40 °C. The diffusion pump oil (Santovac 5, Santovac fluids, Spartanburg, SC, USA) was used between the copper plate and the device to ensure efficient thermal contact. The gas inlet port on the device was connected to the gas sample bag and the gas outlet was connected to a miniature air pump (SKC Inc., 210-1002A). The pump provided negative pressure to the gas outlet of the device to pull the gas from the sample bag in to the device at a controlled flow rate (40–200 mL min
−1). At the end of gas sampling, the capture liquid (approximately 30 μL) was collected from the device using a gas-tight syringe. The VOC content of the collected liquid was analysed by GC-MS. For analysis, the 30 µL liquid samples were injected into a sealed 10 mL GC vial and immediately placed in the headspace sampler for analysis. For each trace, the peak areas were calculated using Chemstation software (Agilent Technologies, Santa Clara, CA, USA).
2.8. Fluoresence Measurements
To quantify the capture liquid collection efficiency, a water soluble fluorescent stain was used as a marker. A calcein (CAS 1461-15-10) stock solution was prepared in water to a concentration of 78 µM, filtered through a 0.22 µm pore size PTFE syringe membrane filter. This solution was loaded to the device and collected to measure the amount of calcein. The plate reader (Enspire 2300 multilabel reader, Perkin Elmer, Waltham, MA, USA) was used to measure fluorescence emission from liquid samples collected from the device. For 96-well plate measurements, the 30 µL collected volume was increased to 100 µL by adding 70 µL of water. The measured emission from the well was converted to calcein amount (moles) by using the calibration curves generated with the standard samples.
4. Conclusions
This study demonstrated the use of an open micro-channel device which can be used as a miniature gas-sampling interface for VOC extraction. The device design utilises capillary flow for capture liquid loading and collection. This is an effective and simple way to charge, sample and recharge a gas-to-liquid extraction device. It was found that maintaining the surface wettability was the key factor in enabling the stable operation of the device. The factors influencing the VOC extraction efficiency were identified using two lipophilic model volatiles with low water solubility. This study showed that gas sampling temperature, chemical modification of the capture liquid and the type of volatile molecule had significant impacts on the capture efficiency. To develop highly efficient gas-to-liquid extraction systems, the identified factors need to be further optimised taking into consideration the properties of target VOCs. This study used GC-MS for VOC detection. However, in the future, the authors envision that detection can be done on a microfluidic system by mixing the extracted liquid sample directly with the biosensors in solution. The proposed gas-sampling interface should be a useful tool to contribute to the development of miniature VOC sensing systems based on biosensors, paving the way for their use in real life applications.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}