Tissue-Engineered Models for Glaucoma Research


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
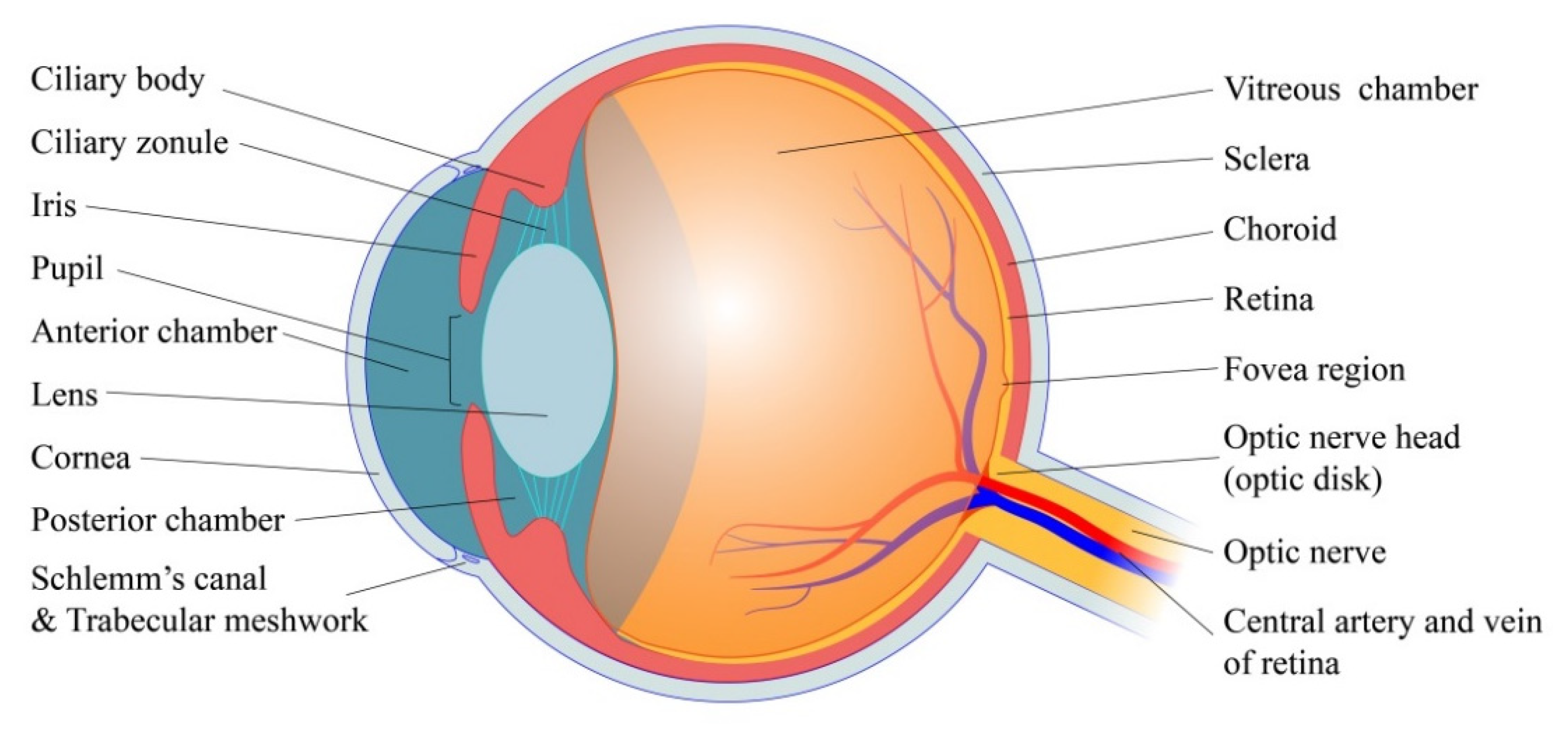
2. Basic Physiology of Eye
2.1. Fluid Dynamics of AH
2.1.1. AH Production
2.1.2. AH Outflow
2.1.3. AH Dynamics
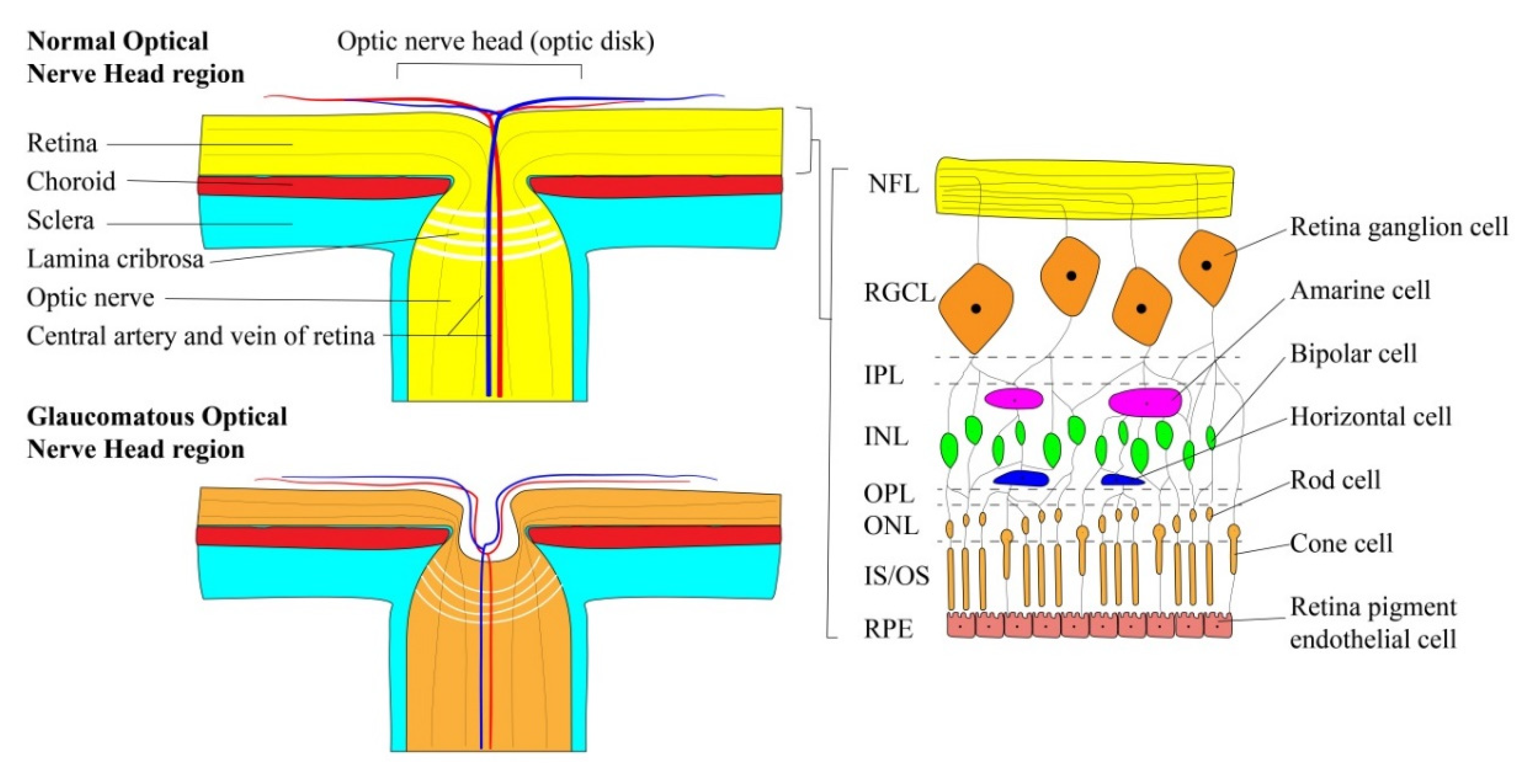
2.2. Retinal Ganglion Cells
3. Glaucoma Pathophysiology
3.1. The Impact of Intraocular Pressure
3.2. Mechanisms of IOP Elevation
4. Trabecular Meshwork and Tissue-Engineered Models
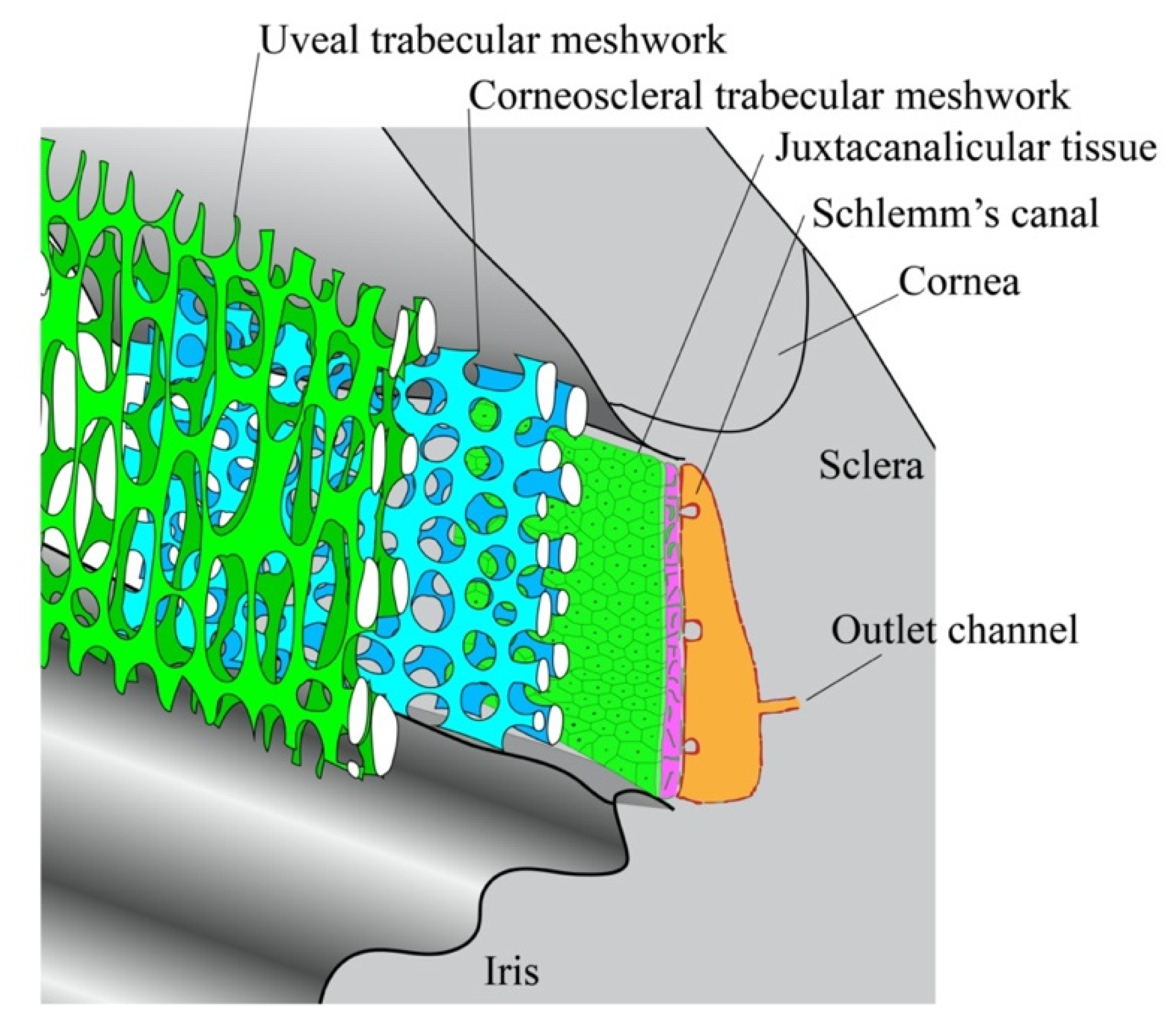
4.1. Substructures in the TM
4.2. Changes of the TM in Glaucoma
4.2.1. Excessive Deposition of ECM in the Pre-Glaucomatous Eye
4.2.2. Changes in Cell Volume Regulation and Cytoskeletal Integrity
4.3. Tissue Engineered Models for Trabecular Pathway Study
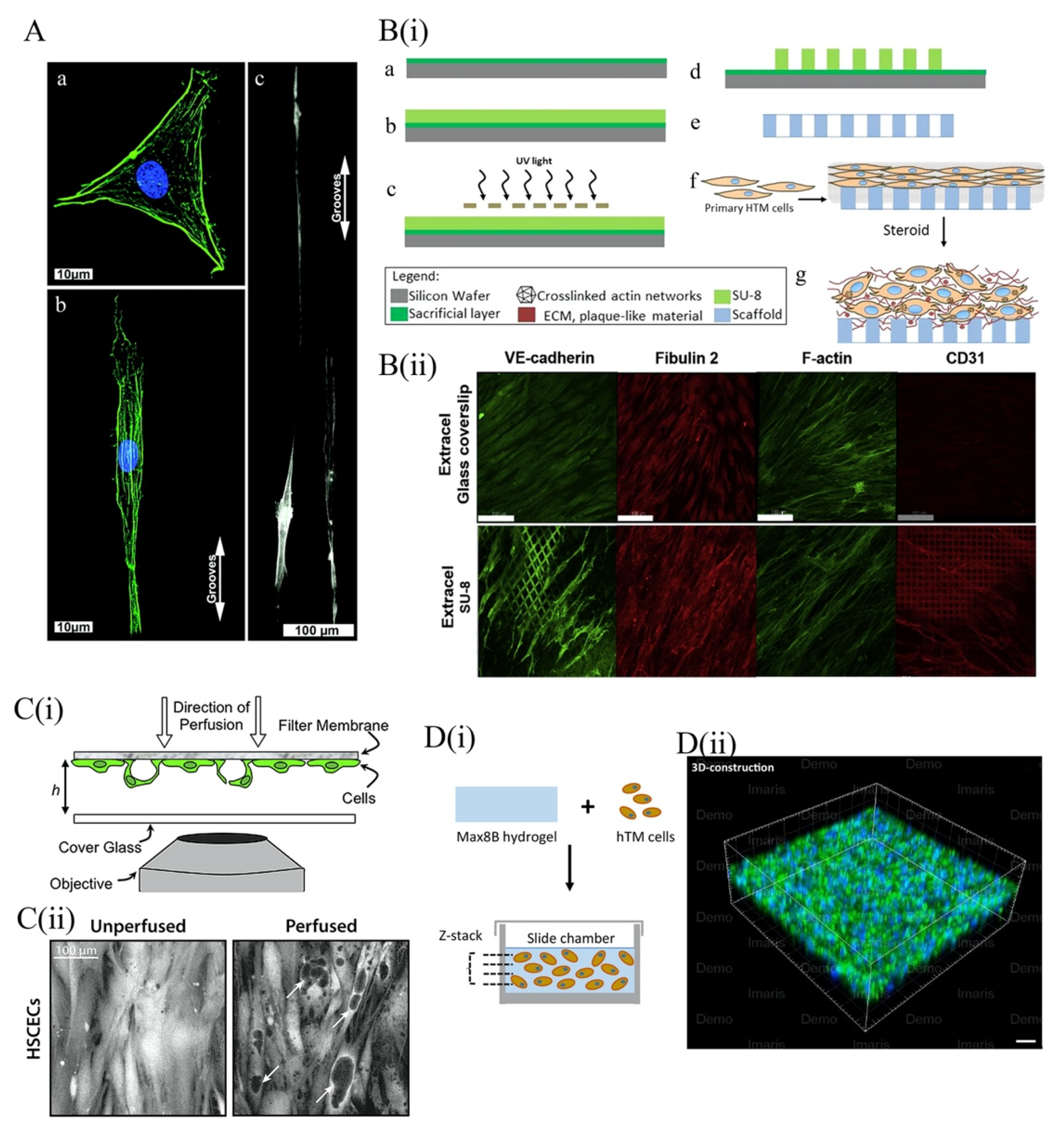
4.3.1. 2D Topographical Scaffold Models of the TM
4.3.2. Three-Dimensional Scaffold for TM Models
5. Tissue-Engineered Models of Retinal Ganglion Cells
5.1. Molecular Mechanisms of Retinal Ganglion Cell Death in Glaucoma
5.1.1. Neurotrophins
5.1.2. Apoptosis Activation
5.2. Tissue-Engineered Models for the Study of Glaucomatous RGCs
5.2.1. Engineered 2D Scaffolds for RGC Culture
5.2.2. Three-Dimensional Hydrogel Scaffolds
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: A review. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreb, R.N.; Khaw, P.T. Primary open-angle glaucoma. Lancet 2004, 363, 1711–1720. [Google Scholar] [CrossRef]
- Tham, Y.-C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.-Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Dai, Y.; Chen, Y.; Yu, D.-Y.; Cringle, S.J.; Chen, J.; Kong, X.; Wang, X.; Jiang, C. Primary angle closure glaucoma: What we know and what we don’t know. Prog. Retin. Eye Res. 2017, 57, 26–45. [Google Scholar] [CrossRef]
- McMonnies, C.W. Glaucoma history and risk factors. J. Optom. 2017, 10, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.K.; Niranjan, D.G.; Agrawal, S.S.; Srivastava, S.; Saxena, R. Recent advances in pharmacotherapy of glaucoma. Indian J. Pharmacol. 2008, 40, 197–208. [Google Scholar] [CrossRef]
- Braunger, B.M.; Fuchshofer, R.; Tamm, E.R. The aqueous humor outflow pathways in glaucoma: A unifying concept of disease mechanisms and causative treatment. Eur. J. Pharm. Biopharm. 2015, 95, 173–181. [Google Scholar] [CrossRef]
- Trivli, A.; Koliarakis, I.; Terzidou, C.; Goulielmos, G.N.; Siganos, C.S.; Spandidos, D.A.; Dalianis, G.; Detorakis, E.T. Normal-tension glaucoma: Pathogenesis and genetics. Exp. Ther. Med. 2019, 17, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Stern, J.H.; Tian, Y.; Funderburgh, J.; Pellegrini, G.; Zhang, K.; Goldberg, J.L.; Ali, R.R.; Young, M.; Xie, Y.; Temple, S. Regenerating Eye Tissues to Preserve and Restore Vision. CellStemCell 2018, 22, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.-Y.; Cringle, S.J.; Morgan, W.H. Glaucoma Related Ocular Structure and Function. In Medical Treatment of Glaucoma; Springer: Singapore, 2019; pp. 1–31. [Google Scholar]
- Goel, M.; Picciani, R.G.; Lee, R.K.; Bhattacharya, S.K. Aqueous humor dynamics: A review. Open Ophthalmol. J. 2010, 4, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, J.P.; Santos, F.M.; Rocha, A.S.; Castro-de-Sousa, J.P.; Queiroz, J.A.; Passarinha, L.A.; Tomaz, C.T. Vitreous humor in the pathologic scope: Insights from proteomic approaches. Proteom. Clin. Appl. 2015, 9, 187–202. [Google Scholar] [CrossRef]
- Remington, L.A. Retina. In Clinical Anatomy and Physiology of the Visual System; Remington, L.A., Ed.; Butterworth-Heinemann: Saint Louis, CA, USA, 2012; pp. 61–92. [Google Scholar]
- Sires, B. Orbital and ocular anatomy. In Textbook of Ophthalmology; Wright, Ed.; Williams and Wilkins: Baltimore, MD, USA, 1997. [Google Scholar]
- Perumal, N.; Manicam, C.; Steinicke, M.; Funke, S.; Pfeiffer, N.; Grus, F.H. Characterization of the human aqueous humour proteome: A comparison of the genders. PLoS ONE 2017, 12, e0172481. [Google Scholar] [CrossRef]
- Delamere, N.A. Ciliary Body and Ciliary Epithelium. Adv. Organ. Biol. 2005, 10, 127–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brubaker, R.F. Measurement of aqueous flow by fluorophotometry. In The Glaucomas; Ritch, R., Shields, M.B., Krupin, T., Eds.; Mosby: St. Louis, MI, USA, 1989; pp. 337–344. [Google Scholar]
- Becker, B. The decline in aqueous secretion and outflow facility with age. Am. J. Ophthalmol. 1958, 46, 731–736. [Google Scholar] [CrossRef]
- Brubaker, R.F. Flow of aqueous humor in humans [The Friedenwald Lecture]. Investig. Ophthalmol. Vis. Sci. 1991, 32, 3145–3166. [Google Scholar]
- Chen, Z.; Sun, J.; Li, M.; Liu, S.; Chen, L.; Jing, S.; Cai, Z.; Xiang, Y.; Song, Y.; Zhang, H.; et al. Effect of age on the morphologies of the human Schlemm’s canal and trabecular meshwork measured with sweptsource optical coherence tomography. Eye 2018, 32, 1621–1628. [Google Scholar] [CrossRef]
- Brubaker, R.F. Measurement with fluorophotometry: I. Plasma binding. II. Anterior segment, and III. Aqueous humor flow. Graefes Arch. Clin. Exp. Ophthalmol. 1985, 222, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Lutjen-Drecoll, E.; Prestele, H.; Rohen, J.W. Structural differences between regions of the ciliary body in primates. Investig. Ophthalmol. Vis. Sci. 1977, 16, 912–924. [Google Scholar]
- Coca-Prados, M.; Sanchez-Torres, J. Molecular approaches to the study of the Na+-K+-ATPase and chloride channels in the ocular ciliary epithelium. In The Eye’s Aqueous Humor; Civan, M.M., Ed.; Academic Press: San Diego, CA, USA, 1998. [Google Scholar]
- Yamaguchi, Y.; Watanabe, T.; Hirakata, A.; Hida, T. Localization and ontogeny of aquaporin-1 and -4 expression in iris and ciliary epithelial cells in rats. Cell Tissue Res. 2006, 325, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Morgan, W.H.; Yu, D.-Y. Mechanism Theories of Glaucoma. In Medical Treatment of Glaucoma; Sun, X., Dai, Y., Eds.; Springer: Singapore, 2019; pp. 33–66. [Google Scholar]
- Bill, A. The role of ciliary blood flow and ultrafiltration in aqueous humor formation. Exp. Eye Res. 1973, 16, 287–298. [Google Scholar] [CrossRef]
- Gabelt, B.T.; Kaufman, P.L. Changes in aqueous humor dynamics with age and glaucoma. Prog. Retin Eye Res. 2005, 24, 612–637. [Google Scholar] [CrossRef] [PubMed]
- Civan, M.M.; Macknight, A.D. The ins and outs of aqueous humour secretion. Exp. Eye Res. 2004, 78, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Li, M. Advanced glaucoma secondary to bilateral idiopathic dilated episcleral veins—A case report. BMC Ophthalmol. 2018, 18, 207. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; McLaren, J.W.; Overby, D.R. Unconventional aqueous humor outflow: A review. Exp. Eye Res. 2017, 158, 94–111. [Google Scholar] [CrossRef] [Green Version]
- Abu-Hassan, D.W.; Acott, T.S.; Kelley, M.J. The Trabecular Meshwork: A Basic Review of Form and Function. J. Ocul. Biol. 2014, 2. [Google Scholar] [CrossRef]
- Bentley, M.D.; Hann, C.R.; Fautsch, M.P. Anatomical Variation of Human Collector Channel Orifices. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1153–1159. [Google Scholar] [CrossRef]
- Johnstone, M.; Martin, E.; Jamil, A. Pulsatile flow into the aqueous veins: Manifestations in normal and glaucomatous eyes. Exp. Eye Res. 2011, 92, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, S.F. The uveoscleral outflow routes. Eye 1997, 11 Pt 2, 149–154. [Google Scholar] [CrossRef]
- Winkler, N.S.; Fautsch, M.P. Effects of prostaglandin analogues on aqueous humor outflow pathways. J. Ocul. Pharmacol. Ther. 2014, 30, 102–109. [Google Scholar] [CrossRef] [Green Version]
- WuDunn, D. Mechanobiology of trabecular meshwork cells. Exp. Eye Res. 2009, 88, 718–723. [Google Scholar] [CrossRef]
- Lei, Y.; Overby, D.R.; Boussommier-Calleja, A.; Stamer, W.D.; Ethier, C.R. Outflow physiology of the mouse eye: Pressure dependence and washout. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1865–1871. [Google Scholar] [CrossRef] [Green Version]
- Klein, B.E.; Klein, R.; Knudtson, M.D. Intraocular pressure and systemic blood pressure: Longitudinal perspective: The Beaver Dam Eye Study. Br. J. Ophthalmol. 2005, 89, 284–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanes, J.R.; Masland, R.H. The types of retinal ganglion cells: Current status and implications for neuronal classification. Annu. Rev. Neurosci. 2015, 38, 221–246. [Google Scholar] [CrossRef]
- Knighton, R.W.; Gregori, G. The shape of the ganglion cell plus inner plexiform layers of the normal human macula. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7412–7420. [Google Scholar] [CrossRef]
- Nelson, R. Visual Responses of Ganglion Cells. In Webvision: The Organization of the Retina and Visual System; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 2001. [Google Scholar]
- Goetz, C.G. Textbook of Clinical Neurology; Saunders: Philadelphia, PA, USA, 2003; pp. 511–529. [Google Scholar]
- Abramoff, M.D.; Garvin, M.K.; Sonka, M. Retinal imaging and image analysis. IEEE Rev. Biomed. Eng. 2010, 3, 169–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruesch, S.R.; Arey, L.B. The number of myelinated and unmyelinated fibers in the optic nerve of vertebrates. J. Comp. Neurol. 1942, 77, 631–665. [Google Scholar] [CrossRef]
- Country, M.W. Retinal metabolism: A comparative look at energetics in the retina. Brain Res. 2017, 1672, 50–57. [Google Scholar] [CrossRef]
- Hein, T.W.; Rosa, R.H., Jr.; Yuan, Z.; Roberts, E.; Kuo, L. Divergent roles of nitric oxide and rho kinase in vasomotor regulation of human retinal arterioles. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1583–1590. [Google Scholar] [CrossRef]
- Bill, A.; Sperber, G.O. Control of retinal and choroidal blood flow. Eye 1990, 4 Pt 2, 319–325. [Google Scholar] [CrossRef]
- Kur, J.; Newman, E.A.; Chan-Ling, T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog. Retin. Eye Res. 2012, 31, 377–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkins, D.J. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog. Retin. Eye Res. 2012, 31, 702–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killer, H.E.; Jaggi, G.P.; Flammer, J.; Miller, N.R.; Huber, A.R. The optic nerve: A new window into cerebrospinal fluid composition? Brain 2006, 129, 1027–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flammer, J.; Orgul, S.; Costa, V.P.; Orzalesi, N.; Krieglstein, G.K.; Serra, L.M.; Renard, J.P.; Stefansson, E. The impact of ocular blood flow in glaucoma. Prog. Retin. Eye Res. 2002, 21, 359–393. [Google Scholar] [CrossRef]
- Guy, A.H.; Wiggs, J.L.; Turalba, A.; Pasquale, L.R. Translating the Low Translaminar Cribrosa Pressure Gradient Hypothesis into the Clinical Care of Glaucoma. Semin. Ophthalmol. 2016, 31, 131–139. [Google Scholar] [CrossRef]
- Berdahl, J.P.; Fautsch, M.P.; Stinnett, S.S.; Allingham, R.R. Intracranial pressure in primary open angle glaucoma, normal tension glaucoma, and ocular hypertension: A case-control study. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5412–5418. [Google Scholar] [CrossRef]
- Ochs, S. Energy metabolism and supply of VP to the fast axoplasmic transport mechanism in nerve. Fed. Proc. 1974, 33, 1049–1058. [Google Scholar]
- Raza, A.S.; Cho, J.; de Moraes, C.G.; Wang, M.; Zhang, X.; Kardon, R.H.; Liebmann, J.M.; Ritch, R.; Hood, D.C. Retinal ganglion cell layer thickness and local visual field sensitivity in glaucoma. Arch. Ophthalmol. 2011, 129, 1529–1536. [Google Scholar] [CrossRef] [Green Version]
- Alexander, K.L. Glaucomatous cupping--appearance, pathogenesis, detection. J. Am. Optom. Assoc. 1978, 49, 1056–1059. [Google Scholar]
- Carpel, E.F.; Engstrom, P.F. The normal cup-disk ratio. Am. J. Ophthalmol. 1981, 91, 588–597. [Google Scholar] [CrossRef]
- Healey, P.R.; Mitchell, P.; Smith, W.; Wang, J.J. Optic disc hemorrhages in a population with and without signs of glaucoma. Ophthalmology 1998, 105, 216–223. [Google Scholar] [CrossRef]
- Elkington, A.R.; Inman, C.B.; Steart, P.V.; Weller, R.O. The structure of the lamina cribrosa of the human eye: An immunocytochemical and electron microscopical study. Eye 1990, 4 Pt 1, 42–57. [Google Scholar] [CrossRef]
- Bristow, E.A.; Griffiths, P.G.; Andrews, R.M.; Johnson, M.A.; Turnbull, D.M. The distribution of mitochondrial activity in relation to optic nerve structure. Arch. Ophthalmol. 2002, 120, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Triviño, A.; Ramírez, J.M.; Salazar, J.J.; Ramírez, A.I. Astroglial Architecture of the Human Optic Nerve: Functional Role of Astrocytes. In Understanding Glial Cells; Castellano, B., González, B., Nieto-Sampedro, M., Eds.; Springer US: Boston, MA, USA, 1998; pp. 63–77. [Google Scholar]
- Trivino, A.; Ramirez, J.M.; Salazar, J.J.; Ramirez, A.I.; Garcia-Sanchez, J. Immunohistochemical study of human optic nerve head astroglia. Vision Res. 1996, 36, 2015–2028. [Google Scholar] [CrossRef] [Green Version]
- Salazar, J.I.; Ramírez, A.; De Hoz, R.; Salobrar-Garcia, E.; Rojas, P.A.; Fernández-Albarral, J.; López-Cuenca, I.; Rojas, B.; Triviño, A.M.; Ramírez, J. Anatomy of the Human Optic Nerve: Structure and Function. In Optic Nerve; Ferreri, F.M., Ed.; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar]
- Lee, S.H.; Kwak, S.W.; Kang, E.M.; Kim, G.A.; Lee, S.Y.; Bae, H.W.; Seong, G.J.; Kim, C.Y. Estimated Trans-Lamina Cribrosa Pressure Differences in Low-Teen and High-Teen Intraocular Pressure Normal Tension Glaucoma: The Korean National Health and Nutrition Examination Survey. PLoS ONE 2016, 11, e0148412. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J.B.; Nangia, V.; Wang, N.; Bhate, K.; Nangia, P.; Nangia, P.; Yang, D.; Xie, X.; Panda-Jonas, S. Trans-lamina cribrosa pressure difference and open-angle glaucoma. The central India eye and medical study. PLoS ONE 2013, 8, e82284. [Google Scholar] [CrossRef] [Green Version]
- Morgan, W.H.; Yu, D.Y.; Alder, V.A.; Cringle, S.J.; Cooper, R.L.; House, P.H.; Constable, I.J. The correlation between cerebrospinal fluid pressure and retrolaminar tissue pressure. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1419–1428. [Google Scholar]
- Morgan, W.H.; Yu, D.Y.; Cooper, R.L.; Alder, V.A.; Cringle, S.J.; Constable, I.J. The influence of cerebrospinal fluid pressure on the lamina cribrosa tissue pressure gradient. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1163–1172. [Google Scholar]
- McMonnies, C.W. The interaction between intracranial pressure, intraocular pressure and lamina cribrosal compression in glaucoma. Clin. Exp. Optom. 2016, 99, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Johannesson, G.; Eklund, A.; Linden, C. Intracranial and Intraocular Pressure at the Lamina Cribrosa: Gradient Effects. Curr. Neurol. Neurosci. Rep. 2018, 18, 25. [Google Scholar] [CrossRef] [Green Version]
- Quigley, H.A.; Addicks, E.M. Regional differences in the structure of the lamina cribrosa and their relation to glaucomatous optic nerve damage. Arch. Ophthalmol. 1981, 99, 137–143. [Google Scholar] [CrossRef]
- Downs, J.C.; Roberts, M.D.; Sigal, I.A. Glaucomatous cupping of the lamina cribrosa: A review of the evidence for active progressive remodeling as a mechanism. Exp. Eye Res. 2011, 93, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, J.B.; Berenshtein, E.; Holbach, L. Anatomic relationship between lamina cribrosa, intraocular space, and cerebrospinal fluid space. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5189–5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.J.; Lu, P.; Zhang, W.F.; Lu, J.H. High myopia as a risk factor in primary open angle glaucoma. Int. J. Ophthalmol. 2012, 5, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Dichtl, A.; Jonas, J.B.; Naumann, G.O. Histomorphometry of the optic disc in highly myopic eyes with absolute secondary angle closure glaucoma. Br. J. Ophthalmol. 1998, 82, 286–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, H.A.; Addicks, E.M.; Green, W.R.; Maumenee, A.E. Optic nerve damage in human glaucoma. II. The site of injury and susceptibility to damage. Arch. Ophthalmol. 1981, 99, 635–649. [Google Scholar] [CrossRef]
- Yu, D.Y.; Cringle, S.J.; Morgan, W.H. Mechanism Theories of Glaucoma In Medical Treatment of Glaucoma; Springer: Singapore, 2019; p. 33. [Google Scholar]
- Park, B.C.; Shen, X.; Samaraweera, M.; Yue, B.Y. Studies of optineurin, a glaucoma gene: Golgi fragmentation and cell death from overexpression of wild-type and mutant optineurin in two ocular cell types. Am. J. Pathol. 2006, 169, 1976–1989. [Google Scholar] [CrossRef] [Green Version]
- Phelps, C.D. Angle-closure glaucoma secondary to ciliary body swelling. Arch. Ophthalmol. 1974, 92, 287–290. [Google Scholar] [CrossRef]
- Quigley, H.A. What’s the choroid got to do with angle closure? Arch. Ophthalmol. 2009, 127, 693–694. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Izumchenko, E.; Kanherkar, R.R.; Teka, M.; Cantor, C.; Manaye, K.; Sidransky, D.; West, M.D.; Makarev, E.; Csoka, A.B. Pro-fibrotic pathway activation in trabecular meshwork and lamina cribrosa is the main driving force of glaucoma. Cell Cycle 2016, 15, 1643–1652. [Google Scholar] [CrossRef]
- He, Y.; Leung, K.W.; Zhuo, Y.H.; Ge, J. Pro370Leu mutant myocilin impairs mitochondrial functions in human trabecular meshwork cells. Mol. Vis. 2009, 15, 815–825. [Google Scholar]
- Vranka, J.A.; Kelley, M.J.; Acott, T.S.; Keller, K.E. Extracellular matrix in the trabecular meshwork: Intraocular pressure regulation and dysregulation in glaucoma. Exp. Eye Res. 2015, 133, 112–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, A.D.; Amini, R. Iris Biomechanics. In Biomechanics of the Eye; Roberts, C.J., Dupps, W.J., Downs, J.C., Eds.; Kugler Publications: Amsterdam, The Netherlands, 2018; p. 282. [Google Scholar]
- Lambert, S.R.; Purohit, A.; Superak, H.M.; Lynn, M.J.; Beck, A.D. Long-term risk of glaucoma after congenital cataract surgery. Am. J. Ophthalmol. 2013, 156, 355–361 e352. [Google Scholar] [CrossRef] [Green Version]
- Kalogeropoulos, D.; Sung, V.C. Pathogenesis of Uveitic Glaucoma. J. Curr. Glaucoma Pract. 2018, 12, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Phulke, S.; Kaushik, S.; Kaur, S.; Pandav, S.S. Steroid-induced Glaucoma: An Avoidable Irreversible Blindness. J. Curr. Glaucoma Pract. 2017, 11, 67–72. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Johnson, D.H. Trabecular meshwork phagocytosis in glaucomatous eyes. Ophthalmologica 1997, 211, 147–152. [Google Scholar] [CrossRef]
- Keller, K.E.; Acott, T.S. The Juxtacanalicular Region of Ocular Trabecular Meshwork: A Tissue with a Unique Extracellular Matrix and Specialized Function. J. Ocul. Biol. 2013, 1, 3. [Google Scholar] [PubMed]
- Keller, K.E.; Kelley, M.J.; Acott, T.S. Extracellular matrix gene alternative splicing by trabecular meshwork cells in response to mechanical stretching. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1164–1172. [Google Scholar] [CrossRef]
- Ashok, A.; Kang, M.H.; Wise, A.S.; Pattabiraman, P.; Johnson, W.M.; Lonigro, M.; Ravikumar, R.; Rhee, D.J.; Singh, N. Prion protein modulates endothelial to mesenchyme-like transition in trabecular meshwork cells: Implications for primary open angle glaucoma. Sci. Rep. 2019, 9, 13090. [Google Scholar] [CrossRef] [Green Version]
- Dautriche, C.N.; Szymanski, D.; Kerr, M.; Torrejon, K.Y.; Bergkvist, M.; Xie, Y.; Danias, J.; Stamer, W.D.; Sharfstein, S.T. A biomimetic Schlemm’s canal inner wall: A model to study outflow physiology, glaucoma pathology and high-throughput drug screening. Biomaterials 2015, 65, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.W. Review of the activation of TGF-beta in immunity. J. Leukoc. Biol. 2009, 85, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.M.; Clark, A.F.; Lipson, K.E.; Andrews, D.; Crean, J.K.; O’Brien, C.J. Anti-connective tissue growth factor antibody treatment reduces extracellular matrix production in trabecular meshwork and lamina cribrosa cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7836–7848. [Google Scholar] [CrossRef] [Green Version]
- Prendes, M.A.; Harris, A.; Wirostko, B.M.; Gerber, A.L.; Siesky, B. The role of transforming growth factor beta in glaucoma and the therapeutic implications. Br. J. Ophthalmol. 2013, 97, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Fleenor, D.L.; Shepard, A.R.; Hellberg, P.E.; Jacobson, N.; Pang, I.H.; Clark, A.F. TGFbeta2-induced changes in human trabecular meshwork: Implications for intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2006, 47, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tellios, N.; Belrose, J.C.; Tokarewicz, A.C.; Hutnik, C.; Liu, H.; Leask, A.; Motolko, M.; Iijima, M.; Parapuram, S.K. TGF-beta induces phosphorylation of phosphatase and tensin homolog: Implications for fibrosis of the trabecular meshwork tissue in glaucoma. Sci. Rep. 2017, 7, 812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Singh, P.; Stamer, W.D.; Singh, D.P. Prdx6 retards senescence and restores trabecular meshwork cell health by regulating reactive oxygen species. Cell Death Discov. 2017, 3, 17060. [Google Scholar] [CrossRef] [Green Version]
- Izzotti, A.; Bagnis, A.; Sacca, S.C. The role of oxidative stress in glaucoma. Mutat. Res. 2006, 612, 105–114. [Google Scholar] [CrossRef]
- Peters, J.C.; Bhattacharya, S.; Clark, A.F.; Zode, G.S. Increased Endoplasmic Reticulum Stress in Human Glaucomatous Trabecular Meshwork Cells and Tissues. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3860–3868. [Google Scholar] [CrossRef] [Green Version]
- Kasetti, R.B.; Maddineni, P.; Millar, J.C.; Clark, A.F.; Zode, G.S. Increased synthesis and deposition of extracellular matrix proteins leads to endoplasmic reticulum stress in the trabecular meshwork. Sci. Rep. 2017, 7, 14951. [Google Scholar] [CrossRef] [Green Version]
- Gasull, X.; Castany, M.; Castellanos, A.; Rezola, M.; Andrés-Bilbé, A.; Canut, M.I.; Estévez, R.; Borrás, T.; Comes, N. The LRRC8-mediated volume-regulated anion channel is altered in glaucoma. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Zhong, Y. Rho/Rho-associated kinase pathway in glaucoma (Review). Int. J. Oncol. 2013, 43, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervan, C.L. Smad-independent TGF-beta2 signaling pathways in human trabecular meshwork cells. Exp. Eye Res. 2017, 158, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Nakao, S.; Arita, R.; Kaizu, Y.; Arima, M.; Zhou, Y.; Kita, T.; Yoshida, S.; Kimura, K.; Isobe, T.; et al. Vascular Normalization by ROCK Inhibitor: Therapeutic Potential of Ripasudil (K-115) Eye Drop in Retinal Angiogenesis and Hypoxia. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2264–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polansky, J.R.; Weinreb, R.N.; Baxter, J.D.; Alvarado, J. Human trabecular cells. I. Establishment in tissue culture and growth characteristics. Investig. Ophthalmol. Vis. Sci. 1979, 18, 1043–1049. [Google Scholar]
- Stone, E.M.; Fingert, J.H.; Alward, W.L.; Nguyen, T.D.; Polansky, J.R.; Sunden, S.L.; Nishimura, D.; Clark, A.F.; Nystuen, A.; Nichols, B.E. Identification of a gene that causes primary open angle glaucoma. Science 1997, 275, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Rybkin, I.; Gerometta, R.; Fridman, G.; Candia, O.; Danias, J. Model systems for the study of steroid-induced IOP elevation. Exp. Eye Res. 2017, 158, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.H. Trabecular meshwork and uveoscleral outflow models. J. Glaucoma 2005, 14, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Tamm, E.R.; Russell, P.; Epstein, D.L.; Johnson, D.H.; Piatigorsky, J. Modulation of myocilin/TIGR expression in human trabecular meshwork. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2577–2582. [Google Scholar]
- Kim, B.; Roberts, C.J.; Mahmoud, A.M.; Weber, P.; Zhao, Y. Effect of topographic cues on in vitro cultured trabecular meshwork endothelial cells. In Proceedings of the 15th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Seattle, WA, USA, 2–6 October 2011; pp. 801–803. [Google Scholar]
- Russell, P.; Gasiorowski, J.Z.; Nealy, P.F.; Murphy, C.J. Response of human trabecular meshwork cells to topographic cues on the nanoscale level. Investig. Ophthalmol. Vis. Sci. 2008, 49, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Kim, B.; Grzybowski, D.; Weber, P.; Roberts, C. Investigation of Microtopography Regulated Human Trabecular Meshwork Culture for Glaucoma Treatment. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4880. [Google Scholar]
- Torrejon, K.Y.; Papke, E.L.; Halman, J.R.; Stolwijk, J.; Dautriche, C.N.; Bergkvist, M.; Danias, J.; Sharfstein, S.T.; Xie, Y. Bioengineered glaucomatous 3D human trabecular meshwork as an in vitro disease model. Biotechnol. Bioeng. 2016, 113, 1357–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrigi, R.M.; Simon, D.; Reed, A.; Stamer, W.D.; Overby, D.R. A model of giant vacuole dynamics in human Schlemm’s canal endothelial cells. Exp. Eye Res. 2011, 92, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waduthanthri, K.D.; He, Y.; Montemagno, C.; Cetinel, S. An injectable peptide hydrogel for reconstruction of the human trabecular meshwork. Acta Biomater. 2019, 100, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Dautriche, C.N.; Xie, Y.; Sharfstein, S.T. Walking through trabecular meshwork biology: Toward engineering design of outflow physiology. Biotechnol. Adv. 2014, 32, 971–983. [Google Scholar] [CrossRef]
- Perkins, T.W.; Alvarado, J.A.; Polansky, J.R.; Stilwell, L.; Maglio, M.; Juster, R. Trabecular meshwork cells grown on filters. Conductivity and cytochalasin effects. Investig. Ophthalmol. Vis. Sci. 1988, 29, 1836–1846. [Google Scholar]
- Torrejon, K.Y.; Papke, E.L.; Halman, J.R.; Bergkvist, M.; Danias, J.; Sharfstein, S.T.; Xie, Y. TGFbeta2-induced outflow alterations in a bioengineered trabecular meshwork are offset by a rho-associated kinase inhibitor. Sci. Rep. 2016, 6, 38319. [Google Scholar] [CrossRef] [Green Version]
- Torrejon, K.Y.; Pu, D.; Bergkvist, M.; Danias, J.; Sharfstein, S.T.; Xie, Y. Recreating a human trabecular meshwork outflow system on microfabricated porous structures. Biotechnol. Bioeng. 2013, 110, 3205–3218. [Google Scholar] [CrossRef]
- Torrejon, K.Y.; Pu, D.; Bergkvist, M.; Sharfstein, S.; Xie, Y.; Tokranova, N.A.; Danias, J. Bioengineering Human Trabecular Meshwork for Glaucoma Therapeutic Screening. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3272. [Google Scholar]
- Tian, Y.I.; Zhang, X.; Torrejon, K.; Danias, J.; Gindina, S.; Nayyar, A.; Du, Y.; Xie, Y. A bioengineering approach to Schlemm’s canal-like stem cell differentiation for in vitro glaucoma drug screening. Acta Biomater. 2020, 105, 203–213. [Google Scholar] [CrossRef]
- Osmond, M.; Bernier, S.M.; Pantcheva, M.B.; Krebs, M.D. Collagen and collagen-chondroitin sulfate scaffolds with uniaxially aligned pores for the biomimetic, three dimensional culture of trabecular meshwork cells. Biotechnol. Bioeng. 2017, 114, 915–923. [Google Scholar] [CrossRef]
- Bouchemi, M.; Roubeix, C.; Kessal, K.; Riancho, L.; Raveu, A.L.; Soualmia, H.; Baudouin, C.; Brignole-Baudouin, F. Effect of benzalkonium chloride on trabecular meshwork cells in a new in vitro 3D trabecular meshwork model for glaucoma. Toxicol. In Vitro 2017, 41, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.H. Human trabecular meshwork cell survival is dependent on perfusion rate. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1204–1208. [Google Scholar]
- McKee, C.T.; Wood, J.A.; Shah, N.M.; Fischer, M.E.; Reilly, C.M.; Murphy, C.J.; Russell, P. The effect of biophysical attributes of the ocular trabecular meshwork associated with glaucoma on the cell response to therapeutic agents. Biomaterials 2011, 32, 2417–2423. [Google Scholar] [CrossRef] [Green Version]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement membrane matrix with biological activity. In Proceedings of Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2005; pp. 378–386. [Google Scholar]
- Huff, R.; Osmond, M.; Krebs, M. In vitro 3D bioprinting trabecular meshwork models using organic hydrogels. Available online: https://mountainscholar.org/handle/11124/171213 (accessed on 23 June 2020).
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Wu, C.L.; Hwang, W.C.; Yang, D.I. More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy. Int. J. Mol. Sci. 2017, 18, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.C.; Guo, Y.; Cepurna, W.O.; Morrison, J.C. Neurotrophin roles in retinal ganglion cell survival: Lessons from rat glaucoma models. Exp. Eye Res. 2009, 88, 808–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, H.A.; McKinnon, S.J.; Zack, D.J.; Pease, M.E.; Kerrigan–Baumrind, L.A.; Kerrigan, D.F.; Mitchell, R.S. Retrograde Axonal Transport of BDNF in Retinal Ganglion Cells Is Blocked by Acute IOP Elevation in Rats. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3460–3466. [Google Scholar]
- Chitranshi, N.; Dheer, Y.; Abbasi, M.; You, Y.; Graham, S.L.; Gupta, V. Glaucoma Pathogenesis and Neurotrophins: Focus on the Molecular and Genetic Basis for Therapeutic Prospects. Curr. Neuropharmacol. 2018, 16, 1018–1035. [Google Scholar] [CrossRef]
- Leaver, S.G.; Cui, Q.; Plant, G.W.; Arulpragasam, A.; Hisheh, S.; Verhaagen, J.; Harvey, A.R. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther. 2006, 13, 1328–1341. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Klöcker, N.; Kermer, P.; Weishaupt, J.H.; Labes, M.; Ankerhold, R.; Bähr, M. Brain-derived neurotrophic factor-mediated neuroprotection of adult rat retinal ganglion cells in vivo does not exclusively depend on phosphatidyl-inositol-3′-kinase/protein kinase B signaling. J. Neurosci. 2000, 20, 6962–6967. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Namekata, K.; Kimura, A.; Harada, C.; Harada, T. ASK1 in neurodegeneration. Adv. Biol. Regul. 2017, 66, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Nickells, R.W.; Semaan, S.J.; Schlamp, C.L. Involvement of the Bcl2 gene family in the signaling and control of retinal ganglion cell death. In Progress in Brain Research; Nucci, C., Cerulli, L., Osborne, N.N., Bagetta, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 173, pp. 423–435. [Google Scholar]
- Wilson, A.M.; Morquette, B.; Feinstein, E.; Di Polo, A. The p53 activators ASPP1 and ASPP2 regulate retinal ganglion cell death in vivo via regulation of Fas and PUMA. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2693. [Google Scholar]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltan, S.; Inman, D.M.; Danilov, C.A.; Morrison, R.S.; Calkins, D.J.; Horner, P.J. Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J. Neurosci. 2010, 30, 5644–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inman, D.M.; Harun-Or-Rashid, M. Metabolic Vulnerability in the Neurodegenerative Disease Glaucoma. Front. Neurosci. 2017, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Li, L.Y.; Patil, R.V.; Wax, M.B. TNF-alpha and TNF-alpha receptor-1 in the retina of normal and glaucomatous eyes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1787–1794. [Google Scholar]
- Tezel, G.; Wax, M.B. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J. Neurosci. 2000, 20, 8693–8700. [Google Scholar] [CrossRef]
- Holler, N.; Tardivel, A.; Kovacsovics-Bankowski, M.; Hertig, S.; Gaide, O.; Martinon, F.; Tinel, A.; Deperthes, D.; Calderara, S.; Schulthess, T.; et al. Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol. Cell. Biol. 2003, 23, 1428–1440. [Google Scholar] [CrossRef] [Green Version]
- Green, D.I.; Ou, Y. Towards the development of a human glaucoma disease-in-a-dish model using stem cells. Expert Rev. Ophthalmol. 2015, 10, 267–280. [Google Scholar] [CrossRef]
- Kador, K.E.; Montero, R.B.; Venugopalan, P.; Hertz, J.; Zindell, A.N.; Valenzuela, D.A.; Uddin, M.S.; Lavik, E.B.; Muller, K.J.; Andreopoulos, F.M.; et al. Tissue engineering the retinal ganglion cell nerve fiber layer. Biomaterials 2013, 34, 4242–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
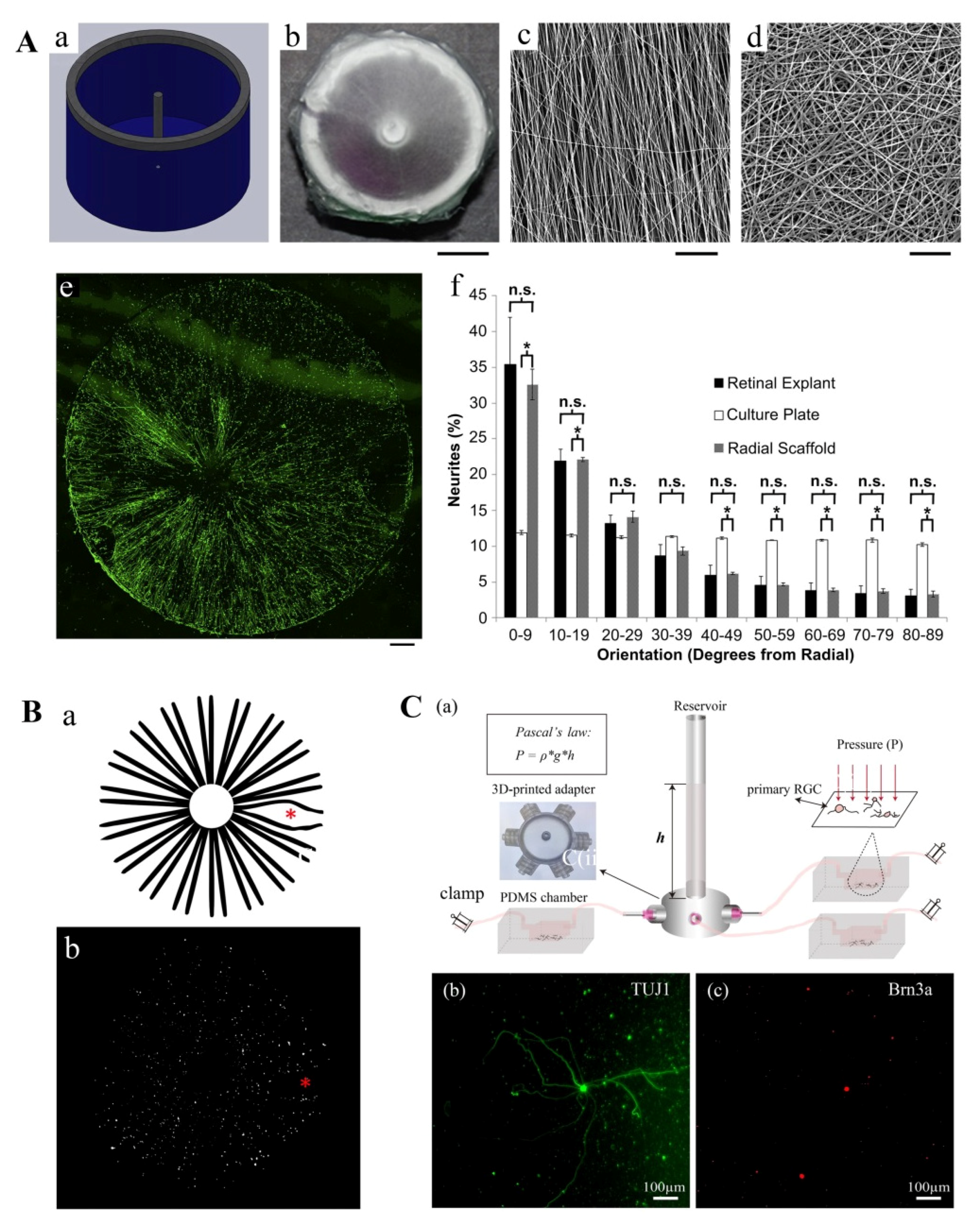
- Kador, K.E.; Grogan, S.P.; Dorthe, E.W.; Venugopalan, P.; Malek, M.F.; Goldberg, J.L.; D’Lima D, D. Control of Retinal Ganglion Cell Positioning and Neurite Growth: Combining 3D Printing with Radial Electrospun Scaffolds. Tissue Eng. Part A 2016, 22, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Mak, H.K.; Chan, Y.K.; Lin, C.; Kong, C.; Leung, C.K.S.; Shum, H.C. An in vitro pressure model towards studying the response of primary retinal ganglion cells to elevated hydrostatic pressures. Sci. Rep. 2019, 9, 9057. [Google Scholar] [CrossRef] [PubMed]
- Kador, K.E.; Alsehli, H.S.; Zindell, A.N.; Lau, L.W.; Andreopoulos, F.M.; Watson, B.D.; Goldberg, J.L. Retinal ganglion cell polarization using immobilized guidance cues on a tissue-engineered scaffold. Acta Biomater. 2014, 10, 4939–4946. [Google Scholar] [CrossRef] [Green Version]
- De Lima, S.; Koriyama, Y.; Kurimoto, T.; Oliveira, J.T.; Yin, Y.; Li, Y.; Gilbert, H.Y.; Fagiolini, M.; Martinez, A.M.; Benowitz, L. Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proc. Natl. Acad. Sci. USA 2012, 109, 9149–9154. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Zhong, X.; Yang, S.; Luo, Z.; Li, K.; Liu, Y.; Cai, S.; Gu, H.; Lu, S.; Zhang, H.; et al. HiPSC-derived retinal ganglion cells grow dendritic arbors and functional axons on a tissue-engineered scaffold. Acta Biomater. 2017, 54, 117–127. [Google Scholar] [CrossRef]
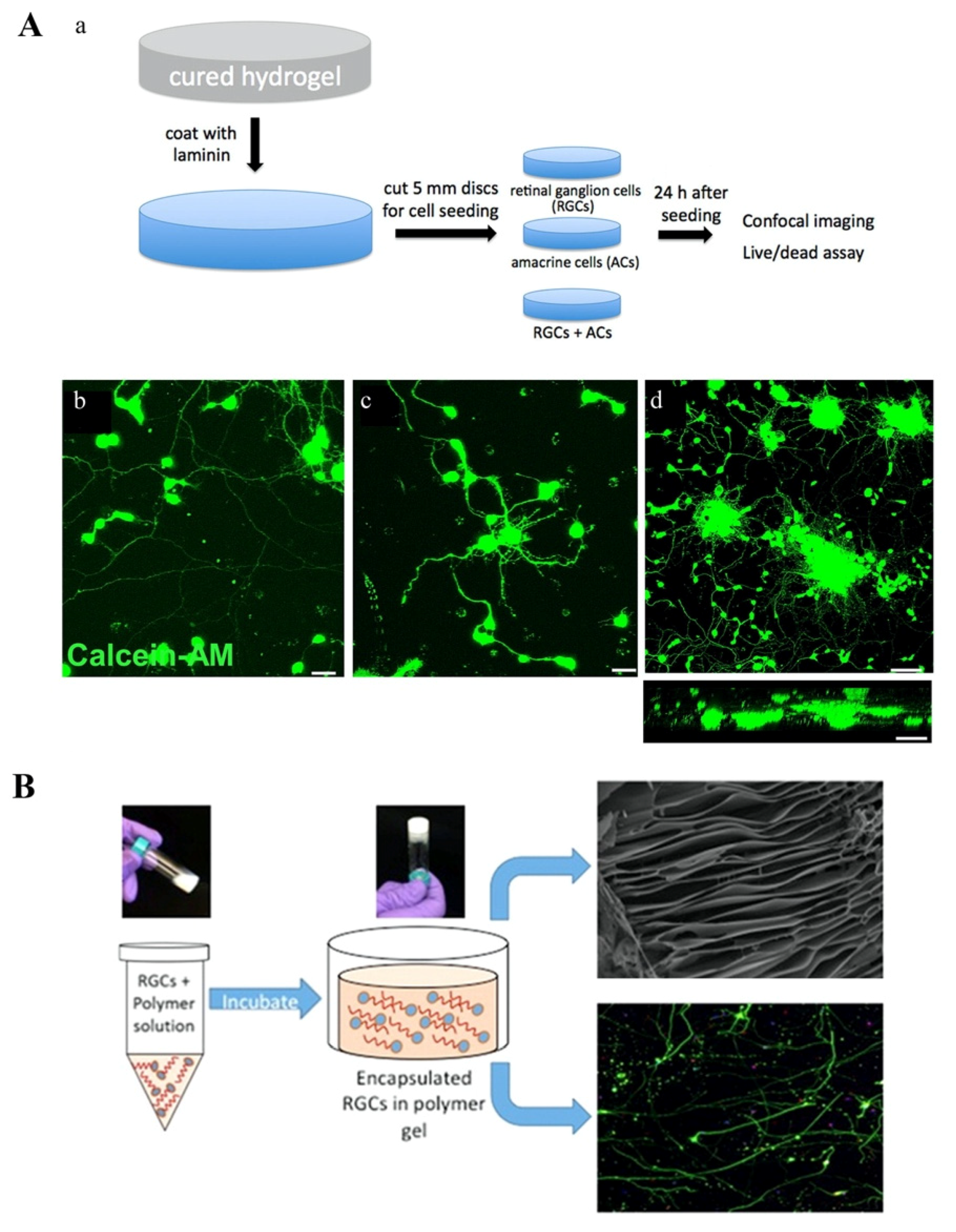
- Hertz, J.; Robinson, R.; Valenzuela, D.A.; Lavik, E.B.; Goldberg, J.L. A tunable synthetic hydrogel system for culture of retinal ganglion cells and amacrine cells. Acta Biomater. 2013, 9, 7622–7629. [Google Scholar] [CrossRef] [Green Version]
- Laughter, M.R.; Ammar, D.A.; Bardill, J.R.; Pena, B.; Kahook, M.Y.; Lee, D.J.; Park, D. A Self-Assembling Injectable Biomimetic Microenvironment Encourages Retinal Ganglion Cell Axon Extension in Vitro. ACS Appl. Mater. Interfaces 2016, 8, 20540–20548. [Google Scholar] [CrossRef] [Green Version]
- Gronthos, S.; Mankani, M.; Brahim, J.; Robey, P.G.; Shi, S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 13625–13630. [Google Scholar] [CrossRef] [Green Version]
- Jagatha, B.; Divya, M.S.; Sanalkumar, R.; Indulekha, C.L.; Vidyanand, S.; Divya, T.S.; Das, A.V.; James, J. In vitro differentiation of retinal ganglion-like cells from embryonic stem cell derived neural progenitors. Biochem. Biophys. Res. Commun. 2009, 380, 230–235. [Google Scholar] [CrossRef]
- Roozafzoon, R.; Lashay, A.; Vasei, M.; Ai, J.; Khoshzaban, A.; Keshel, S.H.; Barabadi, Z.; Bahrami, H. Dental pulp stem cells differentiation into retinal ganglion-like cells in a three dimensional network. Biochem. Biophys. Res. Commun. 2015, 457, 154–160. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, R.; Soden, P.A.; Lee, E. Tissue-Engineered Models for Glaucoma Research. Micromachines 2020, 11, 612. https://doi.org/10.3390/mi11060612
Lu R, Soden PA, Lee E. Tissue-Engineered Models for Glaucoma Research. Micromachines. 2020; 11(6):612. https://doi.org/10.3390/mi11060612
Chicago/Turabian StyleLu, Renhao, Paul A. Soden, and Esak Lee. 2020. "Tissue-Engineered Models for Glaucoma Research" Micromachines 11, no. 6: 612. https://doi.org/10.3390/mi11060612
APA StyleLu, R., Soden, P. A., & Lee, E. (2020). Tissue-Engineered Models for Glaucoma Research. Micromachines, 11(6), 612. https://doi.org/10.3390/mi11060612