
SARS-CoV-2 RNA Detection with Duplex-Specific Nuclease Signal Amplification


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results and Discussion
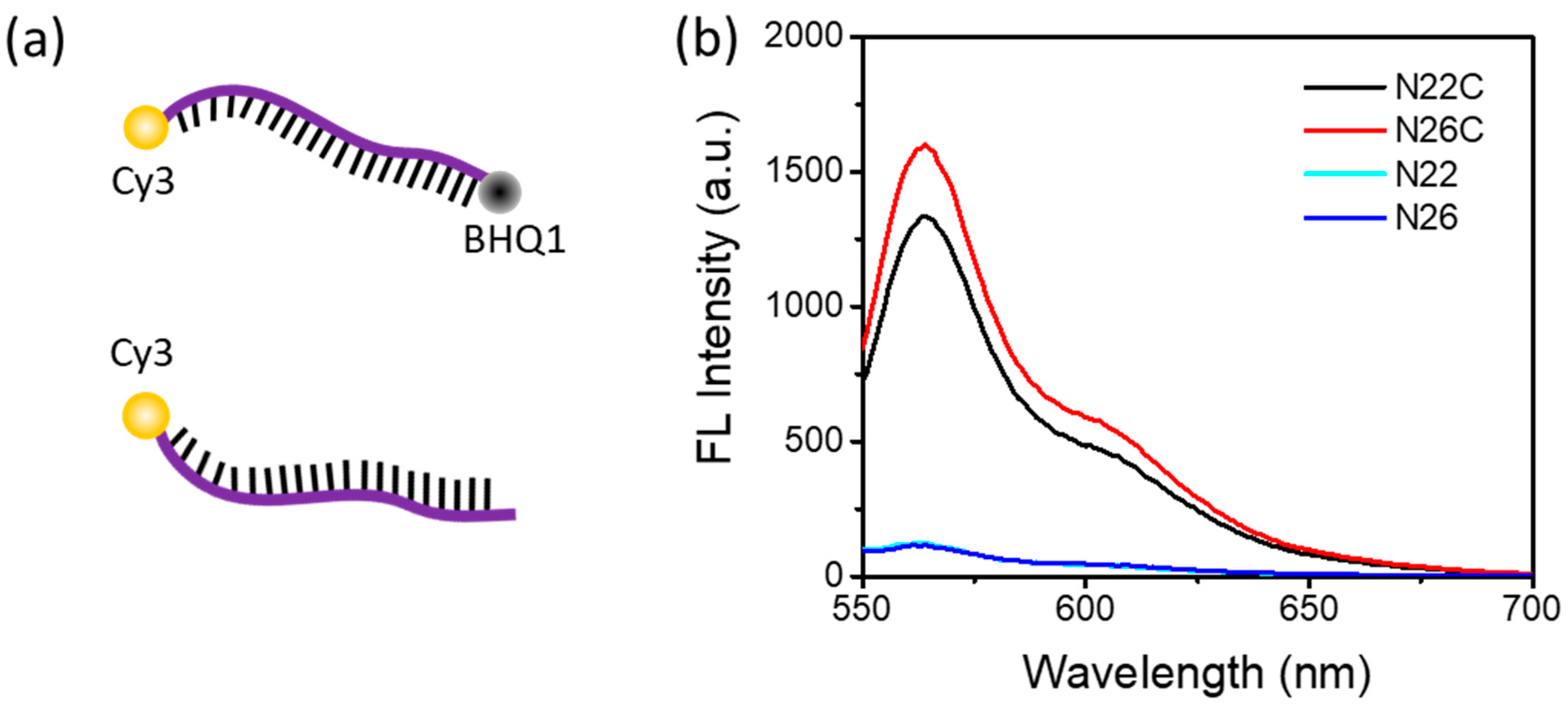
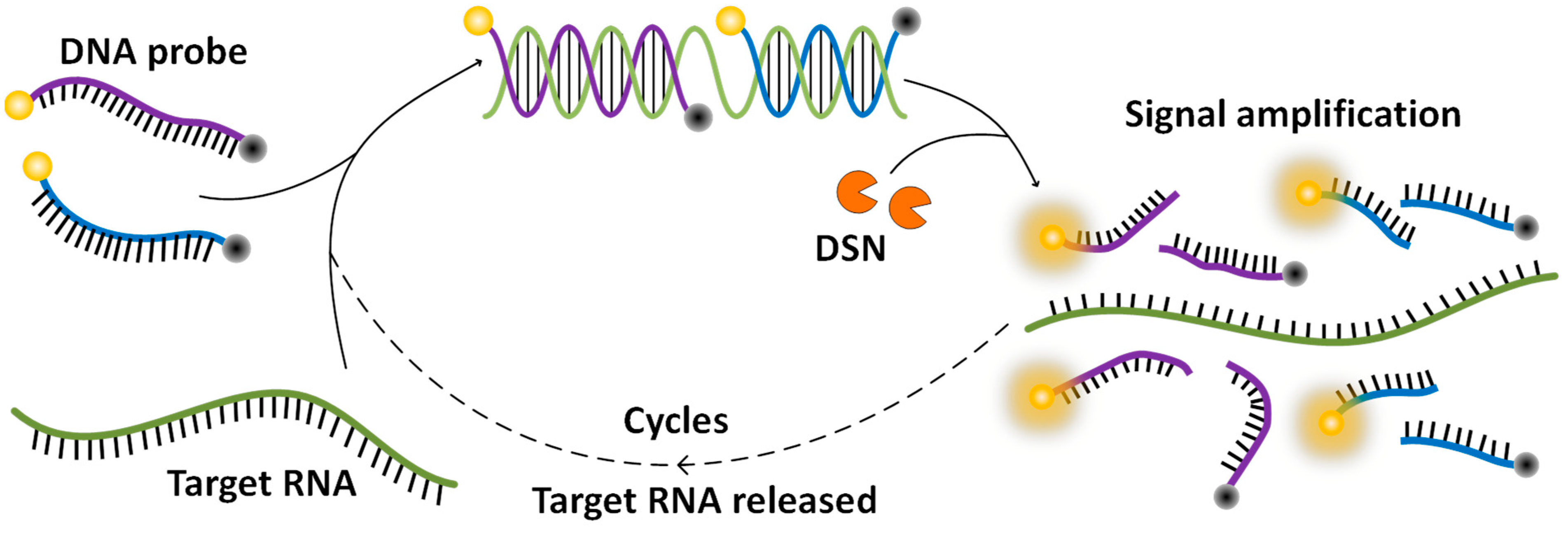
3.1. DNA Probe Design
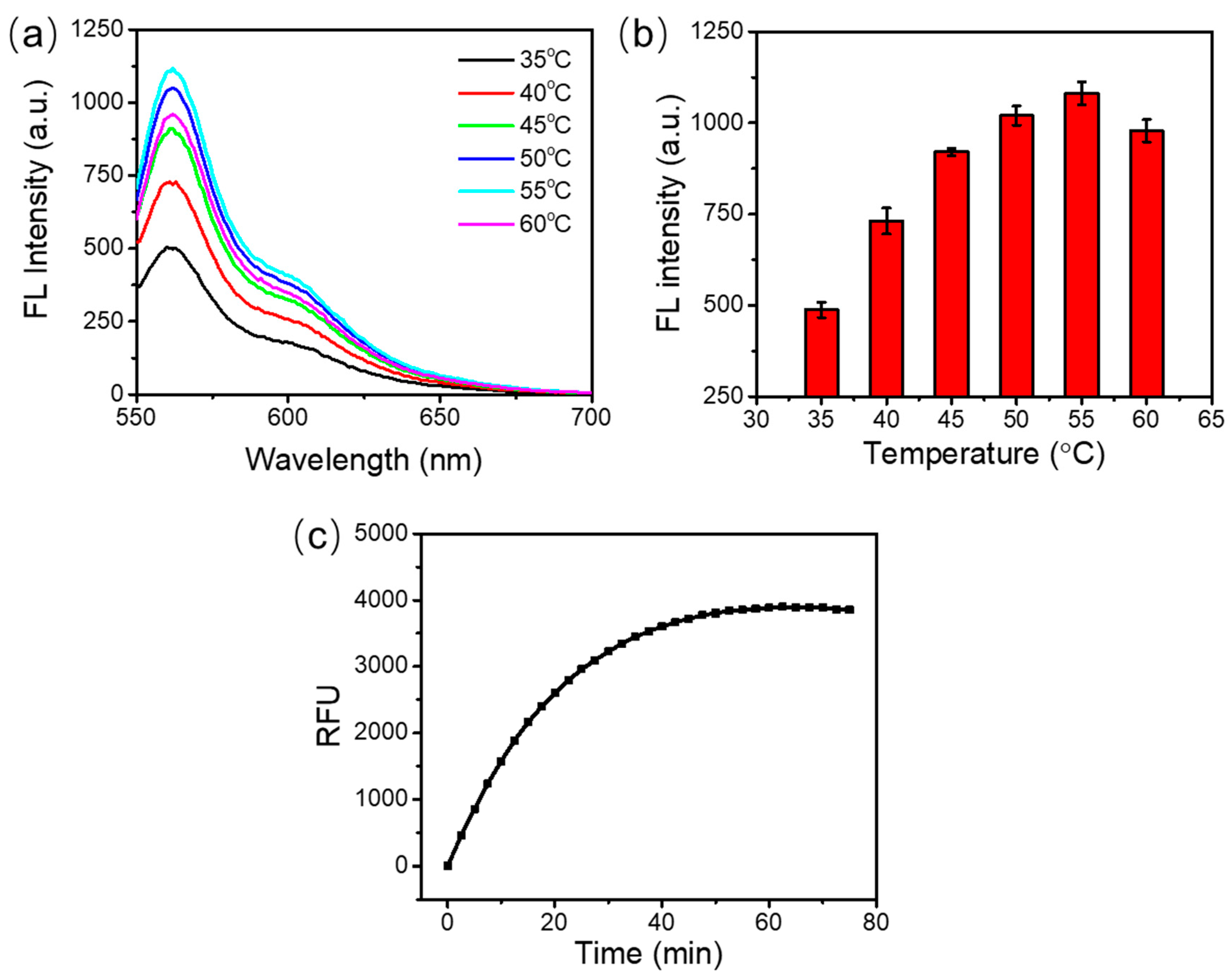
3.2. Optimization of the Incubation Temperature and Incubation Time
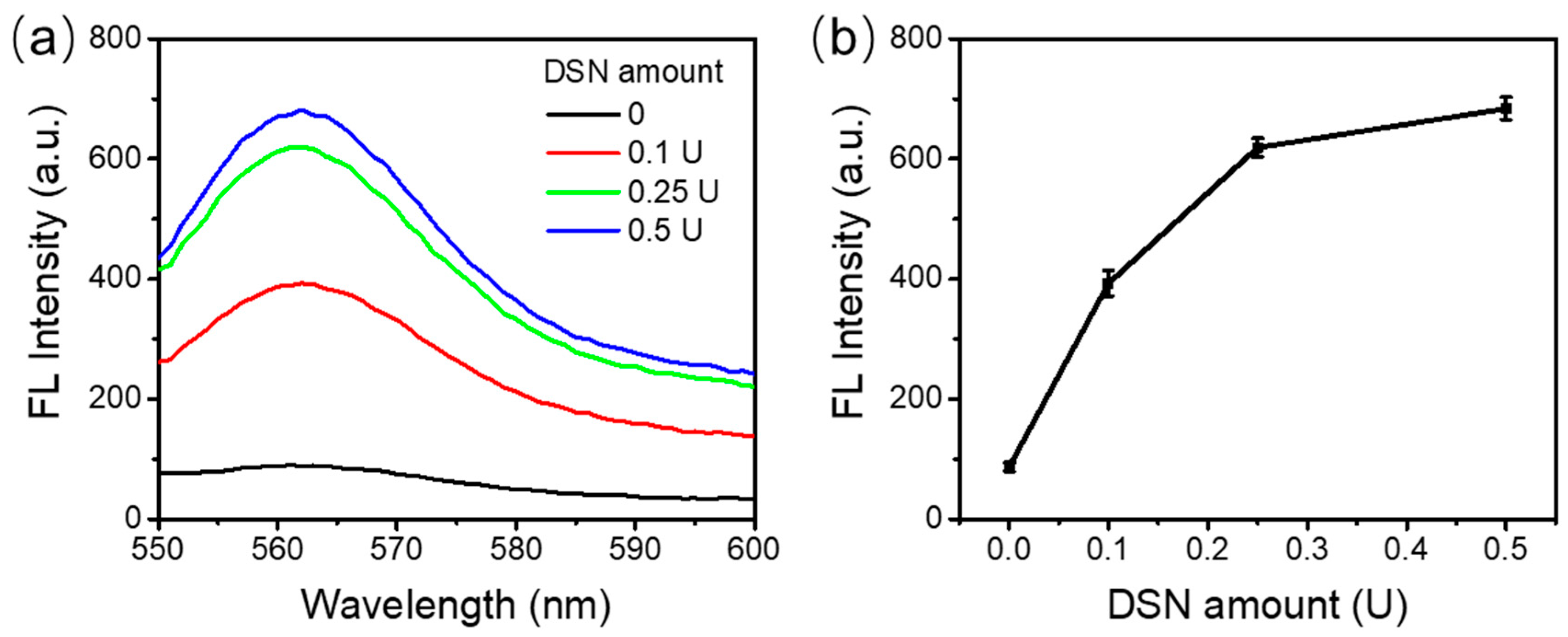
3.3. Optimization of the Amount of DSN Enzyme
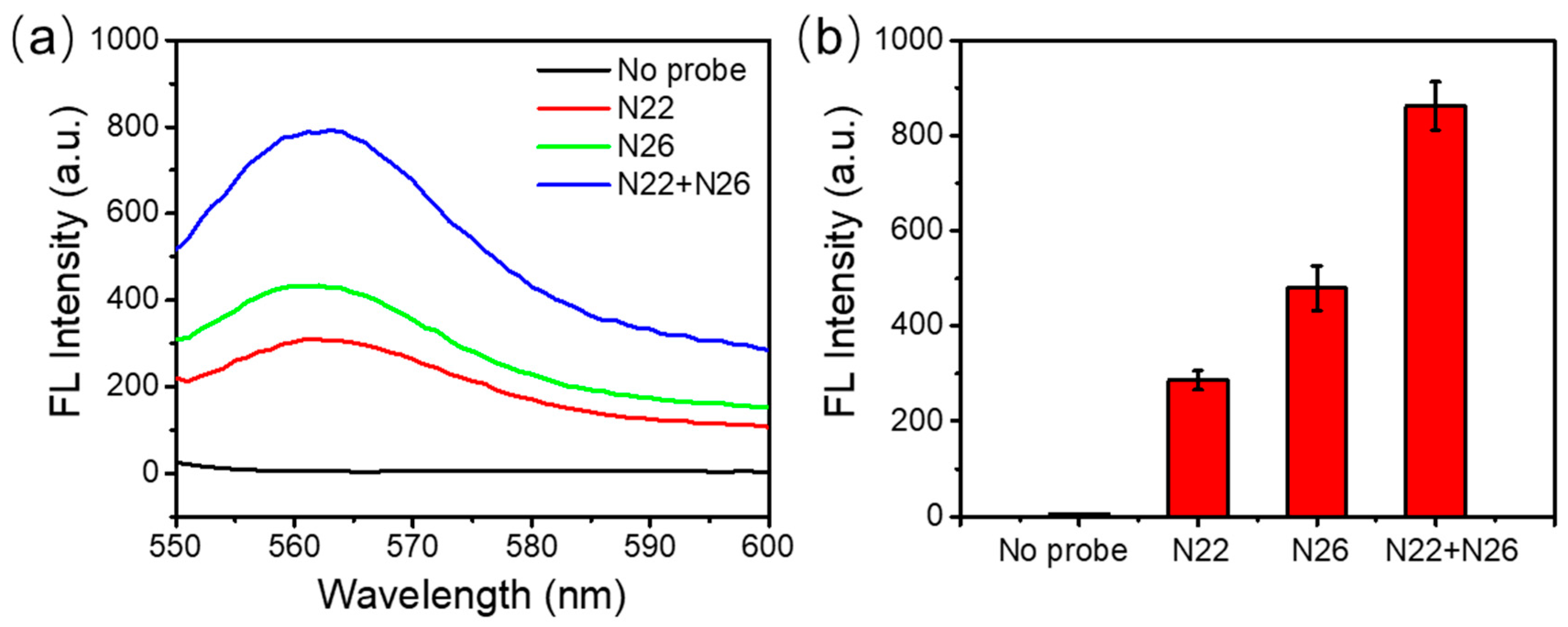
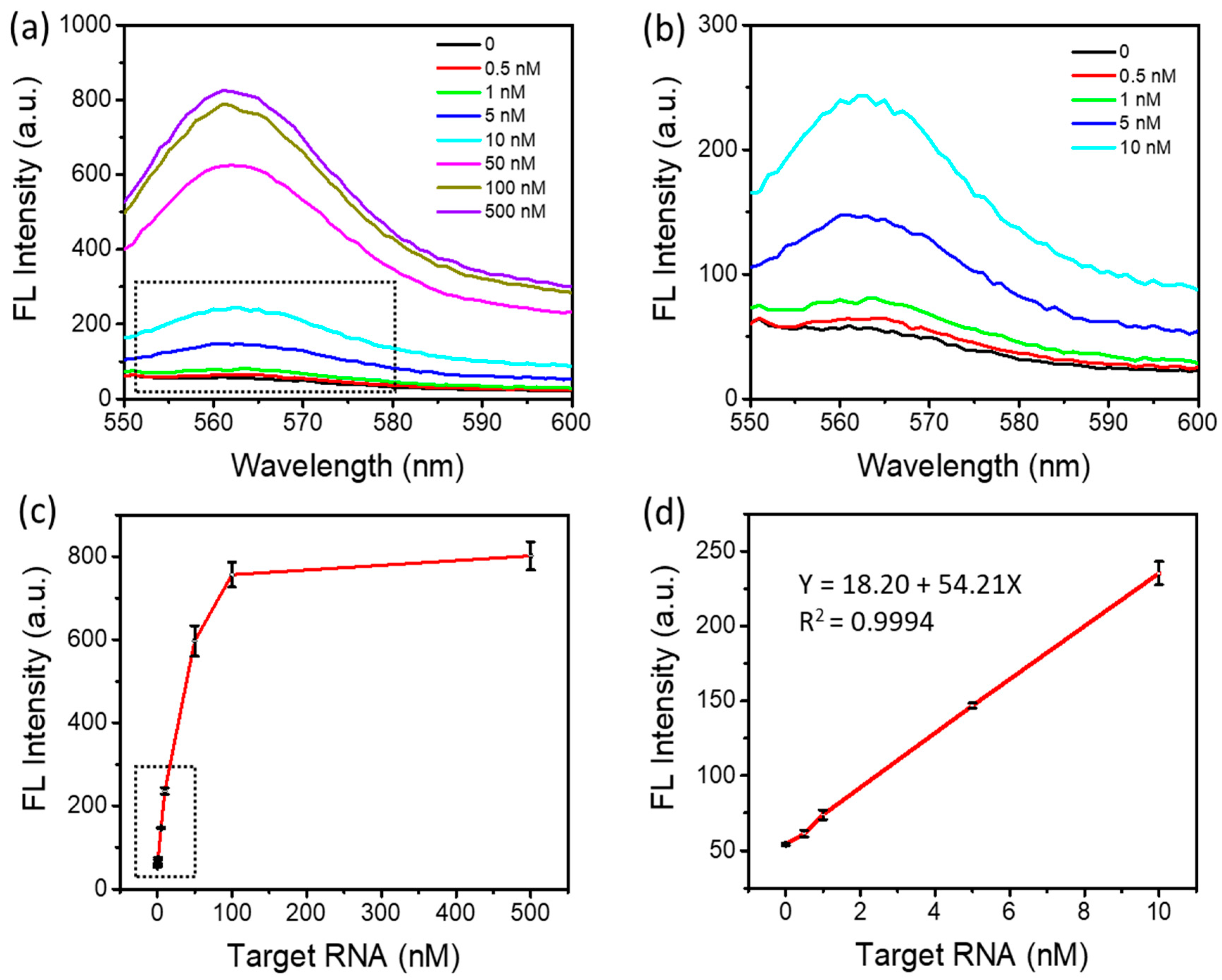
3.4. Enhancement of the Detection Sensitivity and Accuracy
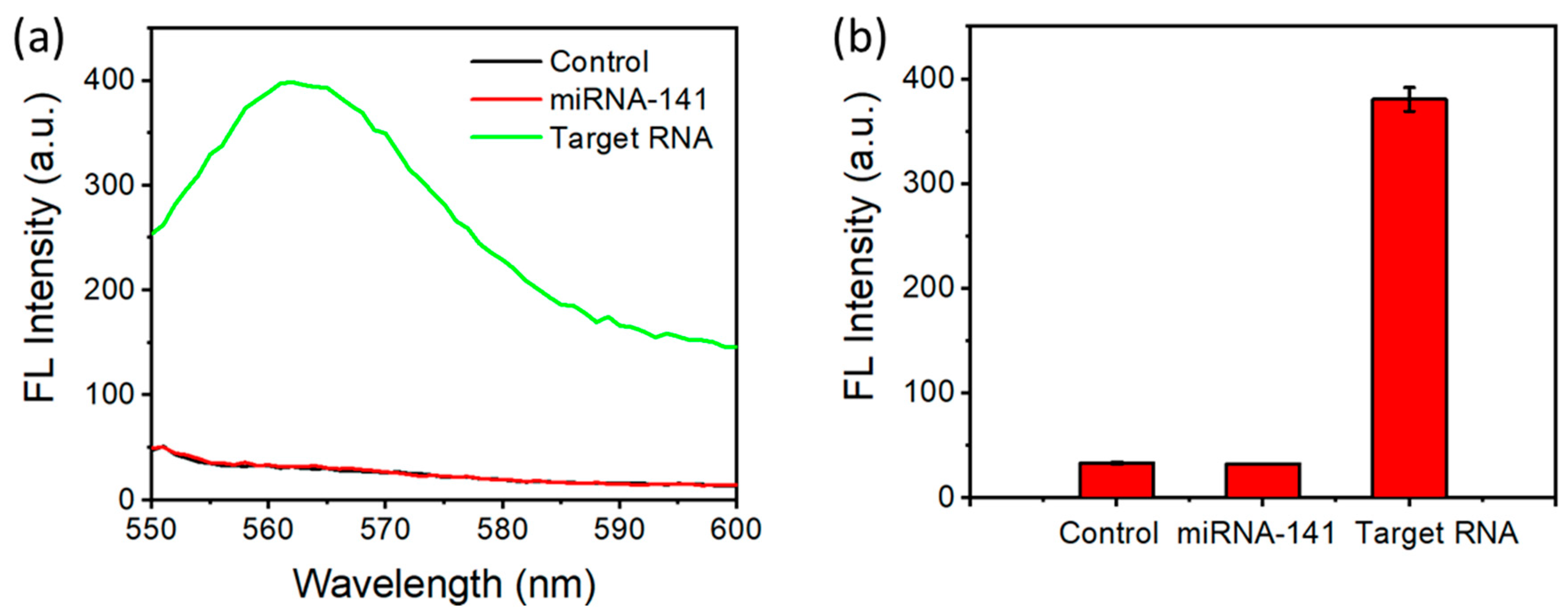
3.5. Detection of SARS-CoV-2 RNA
3.6. Selectivity of SARS-CoV-2 RNA Detection
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nguyen, T.; Bang, D.D.; Wolff, A. 2019 Novel Coronavirus Disease (COVID-19): Paving the Road for Rapid Detection and Point-of-Care Diagnostics. Micromachines 2020, 11, 306. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.L.; Xu, Y.L.; Gao, R.Q.; Lu, R.J.; Han, K.; Wu, G.Z.; Tan, W.J. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA J. Am. Med. Assoc. 2020, 323, 1843–1844. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.W.; Yuan, S.F.; Kok, K.H.; To, K.K.W.; Chu, H.; Yang, J.; Xing, F.F.; Liu, J.L.; Yip, C.C.Y.; Poon, R.W.S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Rothe, C.; Schunk, M.; Sothmann, P.; Bretzel, G.; Froeschl, G.; Wallrauch, C.; Zimmer, T.; Thiel, V.; Janke, C.; Guggemos, W.; et al. Transmission of 2019-nCoV Infection from an Asymptomatic Contact in Germany. N. Engl. J. Med. 2020, 382, 970–971. [Google Scholar] [CrossRef] [Green Version]
- Esbin, M.N.; Whitney, O.N.; Chong, S.S.; Maurer, A.; Darzacq, X.; Tjian, R. Overcoming the bottleneck to widespread testing: A rapid review of nucleic acid testing approaches for COVID-19 detection. Rna 2020, 26, 771–783. [Google Scholar] [CrossRef]
- Giri, B.; Pandey, S.; Shrestha, R.; Pokharel, K.; Ligler, F.S.; Neupane, B.B. Review of analytical performance of COVID-19 detection methods. Anal. Bioanal. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020, 38, 379–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, T.X.; Liu, Z.W.; Wang, G.Q.; Guo, X.G.; Khan, S.A.; Lai, C.C.; Chen, H.Y.; Huang, S.W.; Xia, S.M.; Chen, B.; et al. Detection of COVID-19: A review of the current literature and future perspectives. Biosens. Bioelectron. 2020, 166. [Google Scholar] [CrossRef]
- Khan, M.B.; Zhang, Z.Y.; Li, L.; Zhao, W.; Al Hababi, M.A.M.; Yang, X.D.; Abbasi, Q.H. A Systematic Review of Non-Contact Sensing for Developing a Platform to Contain COVID-19. Micromachines 2020, 11, 912. [Google Scholar] [CrossRef] [PubMed]
- Ravi, N.; Cortade, D.L.; Ng, E.; Wang, S.X. Diagnostics for SARS-CoV-2 detection: A comprehensive review of the FDA-EUA COVID-19 testing landscape. Biosens. Bioelectron. 2020, 165. [Google Scholar] [CrossRef]
- Taha, B.A.; Al Mashhadany, Y.; Mokhtar, M.H.H.; Bin Zan, M.S.D.; Arsad, N. An Analysis Review of Detection Coronavirus Disease 2019 (COVID-19) Based on Biosensor Application. Sensors 2020, 20, 6764. [Google Scholar] [CrossRef]
- Qiu, G.G.; Gai, Z.B.; Tao, Y.L.; Schmitt, J.; Kullak-Ublick, G.A.; Wang, J. Dual-Functional Plasmonic Photothermal Biosensors for Highly Accurate Severe Acute Respiratory Syndrome Coronavirus 2 Detection. ACS Nano 2020, 14, 5268–5277. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.; Lee, G.; Kim, M.J.; Baek, S.H.; Choi, M.; Ku, K.B.; Lee, C.S.; Jun, S.; Park, D.; Kim, H.G.; et al. Rapid Detection of COVID-19 Causative Virus (SARS-CoV-2) in Human Nasopharyngeal Swab Specimens Using Field-Effect Transistor-Based Biosensor (vol 14, pg 5135, 2020). ACS Nano 2020, 14, 12257–12258. [Google Scholar] [CrossRef]
- Ahmadivand, A.; Gerislioglu, B.; Ramezani, Z.; Kaushik, A.; Manickam, P.; Ghoreishi, S.A. Functionalized terahertz plasmonic metasensors: Femtomolar-level detection of SARS-CoV-2 spike proteins. Biosens. Bioelectron. 2021, 177, 112971. [Google Scholar] [CrossRef]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Muller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.W.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, T.; Kojima, S.; Shinohara, M.; Uchida, K.; Fukushi, S.; Hoshino, F.B.; Takeda, N.; Katayama, K. Broadly reactive and highly sensitive assay for Norwalk-like viruses based on real-time quantitative reverse transcription-PCR. J. Clin. Microbiol. 2003, 41, 1548–1557. [Google Scholar] [CrossRef] [Green Version]
- Carter, L.J.; Garner, L.V.; Smoot, J.W.; Li, Y.Z.; Zhou, Q.Q.; Saveson, C.J.; Sasso, J.M.; Gregg, A.C.; Soares, D.J.; Beskid, T.R.; et al. Assay Techniques and Test Development for COVID-19 Diagnosis. ACS Cent. Sci. 2020, 6, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Freeman, W.M.; Walker, S.J.; Vrana, K.E. Quantitative RT-PCR: Pitfalls and potential. Biotechniques 1999, 26, 112–125. [Google Scholar] [CrossRef]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Benvenuto, D.; Giovanetti, M.; Ciccozzi, A.; Spoto, S.; Angeletti, S.; Ciccozzi, M. The 2019-new coronavirus epidemic: Evidence for virus evolution. J. Med. Virol. 2020, 92, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Han, H.; Liu, F.; Lv, Z.H.; Wu, K.L.; Liu, Y.L.; Feng, Y.; Zhu, C.L. Positive rate of RT-PCR detection of SARS-CoV-2 infection in 4880 cases from one hospital in Wuhan, China, from Jan to Feb 2020. Clin. Chim. Acta 2020, 505, 172–175. [Google Scholar] [CrossRef]
- Ai, J.W.; Zhang, Y.; Zhang, H.C.; Xu, T.; Zhang, W.H. Era of molecular diagnosis for pathogen identification of unexplained pneumonia, lessons to be learned. Emerg. Microbes Infec. 2020, 9, 597–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.J.; Xiao, Y.; Kang, L.; Ma, W.T.; Shi, L.S.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.X.; Yang, D.H.; et al. Genomic Diversity of Severe Acute Respiratory Syndrome-Coronavirus 2 in Patients With Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.L.; Wu, C.C.; Li, X.; Song, Y.H.; Yao, X.M.; Wu, X.K.; Duan, Y.G.; Zhang, H.; Wang, Y.R.; Qian, Z.H.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- Shagin, D.A.; Rebrikov, D.V.; Kozhemyako, V.B.; Altshuler, I.M.; Shcheglov, A.S.; Zhulidov, P.A.; Bogdanova, E.A.; Staroverov, D.B.; Rasskazov, V.A.; Lukyanov, S. A novel method for SNP detection using a new duplex-specific nuclease from crab hepatopancreas. Genome Res. 2002, 12, 1935–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.P.; Zhang, H.; Yu, H.L.; Jiang, T.L.; Luo, Y. Duplex-specific nuclease-mediated bioanalysis. Trends Biotechnol. 2015, 33, 180–188. [Google Scholar] [CrossRef]
- Gerasimova, Y.V.; Kolpashchikov, D.M. Enzyme-assisted target recycling (EATR) for nucleic acid detection. Chem. Soc. Rev. 2014, 43, 6405–6438. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.L.; Guo, M.M.; Tang, H.; Wu, Z.; Tang, L.J.; Yu, R.Q.; Jiang, J.H. Nucleic acid amplification-based methods for microRNA detection. Anal. Methods 2015, 7, 2258–2263. [Google Scholar] [CrossRef]
- Qing, T.P.; He, D.G.; He, X.X.; Wang, K.M.; Xu, F.Z.; Wen, L.; Shangguan, J.F.; Mao, Z.G.; Lei, Y.L. Nucleic acid tool enzymes-aided signal amplification strategy for biochemical analysis: Status and challenges. Anal. Bioanal. Chem. 2016, 408, 2793–2811. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Q.; Dong, L.J.; Zhang, J.Y.; Zhao, Y.Q.; Li, Z.P. Recent advances in microRNA detection. Analyst 2018, 143, 1758–1774. [Google Scholar] [CrossRef] [PubMed]
- Zhulidov, P.A.; Bogdanova, E.A.; Shcheglov, A.S.; Vagner, L.L.; Khaspekov, G.L.; Kozhemyako, V.B.; Matz, M.V.; Meleshkevitch, E.; Moroz, L.L.; Lukyanov, S.A.; et al. Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 2004, 32. [Google Scholar] [CrossRef]
- Yin, B.C.; Liu, Y.Q.; Ye, B.C. One-Step, Multiplexed Fluorescence Detection of microRNAs Based on Duplex-Specific Nuclease Signal Amplification. J. Am. Chem. Soc. 2012, 134, 5064–5067. [Google Scholar] [CrossRef]
- Moitra, P.; Alafeef, M.; Dighe, K.; Frieman, M.B.; Pan, D. Selective Naked-Eye Detection of SARS-CoV-2 Mediated by N Gene Targeted Antisense Oligonucleotide Capped Plasmonic Nanoparticles. ACS Nano 2020, 14, 7617–7627. [Google Scholar] [CrossRef] [PubMed]
- Udugama, B.; Kadhiresan, P.; Kozlowski, H.N.; Malekjahani, A.; Osborne, M.; Li, V.Y.C.; Chen, H.M.; Mubareka, S.; Gubbay, J.B.; Chan, W.C.W. Diagnosing COVID-19: The Disease and Tools for Detection. ACS Nano 2020, 14, 3822–3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Xu, X.W.; Li, X.; Zhang, N.; Jiang, W. pH-responsive ZnO nanoprobe mediated DNAzyme signal amplification strategy for sensitive detection and live cell imaging of multiple microRNAs. Sens. Actuators B-Chem. 2019, 293, 93–99. [Google Scholar] [CrossRef]
- Tian, T.; Xiao, H.; Zhang, Z.G.; Long, Y.L.; Peng, S.; Wang, S.R.; Zhou, X.; Liu, S.M.; Zhou, X. Sensitive and Convenient Detection of microRNAs Based on Cascade Amplification by Catalytic DNAzymes. Chem. -a Eur. J. 2013, 19, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.F.C.; Wang, S.S.; Peng, F.; He, J.A.; He, L.; Guo, Y.C.; Gu, D.Y.; Kwok, H.C.; Wu, S.Y.; Ho, H.P.; et al. A non-PCR SPR platform using RNase H to detect MicroRNA 29a-3p from throat swabs of human subjects with influenza A virus H1N1 infection. Analyst 2015, 140, 4566–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chand, R.; Neethirajan, S. Microfluidic platform integrated with graphene-gold nano-composite aptasensor for one-step detection of norovirus. Biosens. Bioelectron. 2017, 98, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Baek, Y.H.; Um, J.; Antigua, K.J.C.; Park, J.H.; Kim, Y.; Oh, S.; Kim, Y.I.; Choi, W.S.; Kim, S.O.Y.; Jeong, J.W.; et al. Development of a reverse transcription-loop-mediated isothermal amplification as a rapid early-detection method for novel SARS-CoV-2. Emerg. Microbes Infec. 2020, 9, 998–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogels, C.B.F.; Brito, A.F.; Wyllie, A.L.; Fauver, J.R.; Ott, I.M.; Kalinich, C.C.; Petrone, M.E.; Casanovas-Massana, A.; Muenker, M.C.; Moore, A.J.; et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat. Microbiol. 2020, 5, 1299–1305. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′-3′) | Melting Temperatures (°C) |
|---|---|---|
| Target RNA | GUAGGGGAACUUCUCCUGCUAGAAUGGCU- GGCAAUGGCGGUGAUGCUGCUCUUGCUU | |
| N22 | Cy3-CAAGAGCAGCATCACCGCCATT-BHQ1 | 70.8 |
| N26 | Cy3-AGCCATTCTAGCAGGAGAAGTTCCCC-BHQ1 | 71.9 |
| N22C | Cy3-CAAGAGCAGCATCACCGCCATT | 70.8 |
| N26C | Cy3-AGCCATTCTAGCAGGAGAAGTTCCCC | 71.9 |
| miRNA-141 | UAACACUGUCUGGUAAAGAUGG |
| Analyte | Linear Range | LOD | References |
|---|---|---|---|
| miRNA-21 (Fluorometric) | 100 pM–30 nM | 54 pM | [39] |
| miRNA-141 (Colorimetric) | 20 pM–10 nM | 20 pM | [40] |
| miRNA-29a-3p (MARS) | 1 nM–70 nM | 1 nM | [41] |
| Norovirus RNA (Microfluidic) | 100 pM–3.5 nM | 100 pM | [42] |
| SARS-CoV-2 RNA (RT-LAMP) | 100 copies/reaction | [43] | |
| SARS-CoV-2 RNA (RT-PCR) | 0.15–100 copies/μL | [16,44] | |
| SARS-CoV-2 RNA | 500 pM–10 nM | 500 pM | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Li, H.; Jia, Y.; Mak, P.-I.; Martins, R.P. SARS-CoV-2 RNA Detection with Duplex-Specific Nuclease Signal Amplification. Micromachines 2021, 12, 197. https://doi.org/10.3390/mi12020197
Liu M, Li H, Jia Y, Mak P-I, Martins RP. SARS-CoV-2 RNA Detection with Duplex-Specific Nuclease Signal Amplification. Micromachines. 2021; 12(2):197. https://doi.org/10.3390/mi12020197
Chicago/Turabian StyleLiu, Meiqing, Haoran Li, Yanwei Jia, Pui-In Mak, and Rui P. Martins. 2021. "SARS-CoV-2 RNA Detection with Duplex-Specific Nuclease Signal Amplification" Micromachines 12, no. 2: 197. https://doi.org/10.3390/mi12020197
APA StyleLiu, M., Li, H., Jia, Y., Mak, P. -I., & Martins, R. P. (2021). SARS-CoV-2 RNA Detection with Duplex-Specific Nuclease Signal Amplification. Micromachines, 12(2), 197. https://doi.org/10.3390/mi12020197