
Cystic Fibrosis Human Organs-on-a-Chip

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
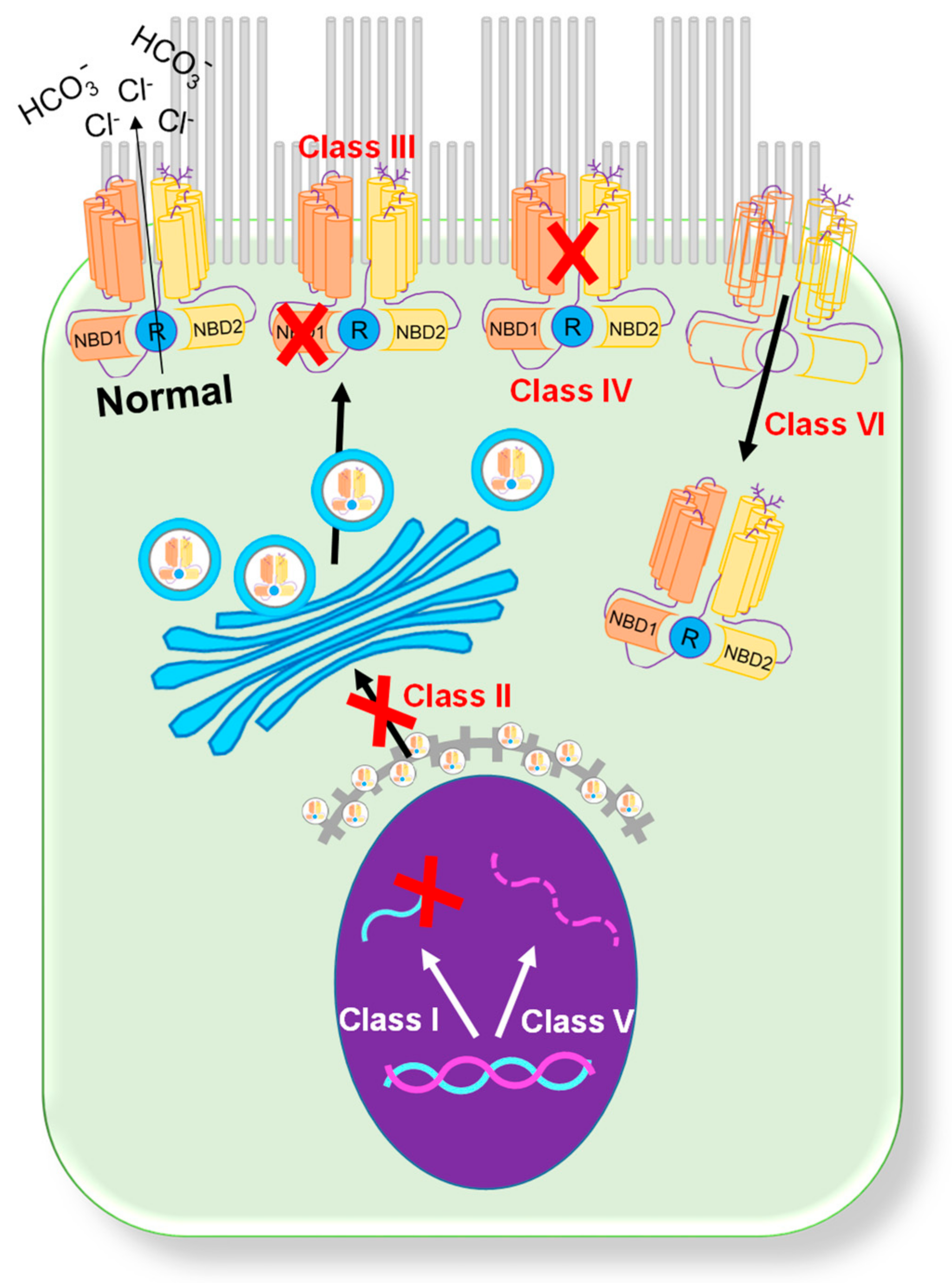
:1. CFTR Mutations
2. Rescue of CFTR Function
2.1. Potentiator Treatment
2.2. Corrector Treatment
2.3. Trikafta Treatment
3. CF Organs
3.1. Pancreas
3.2. Gastrointestinal Tract
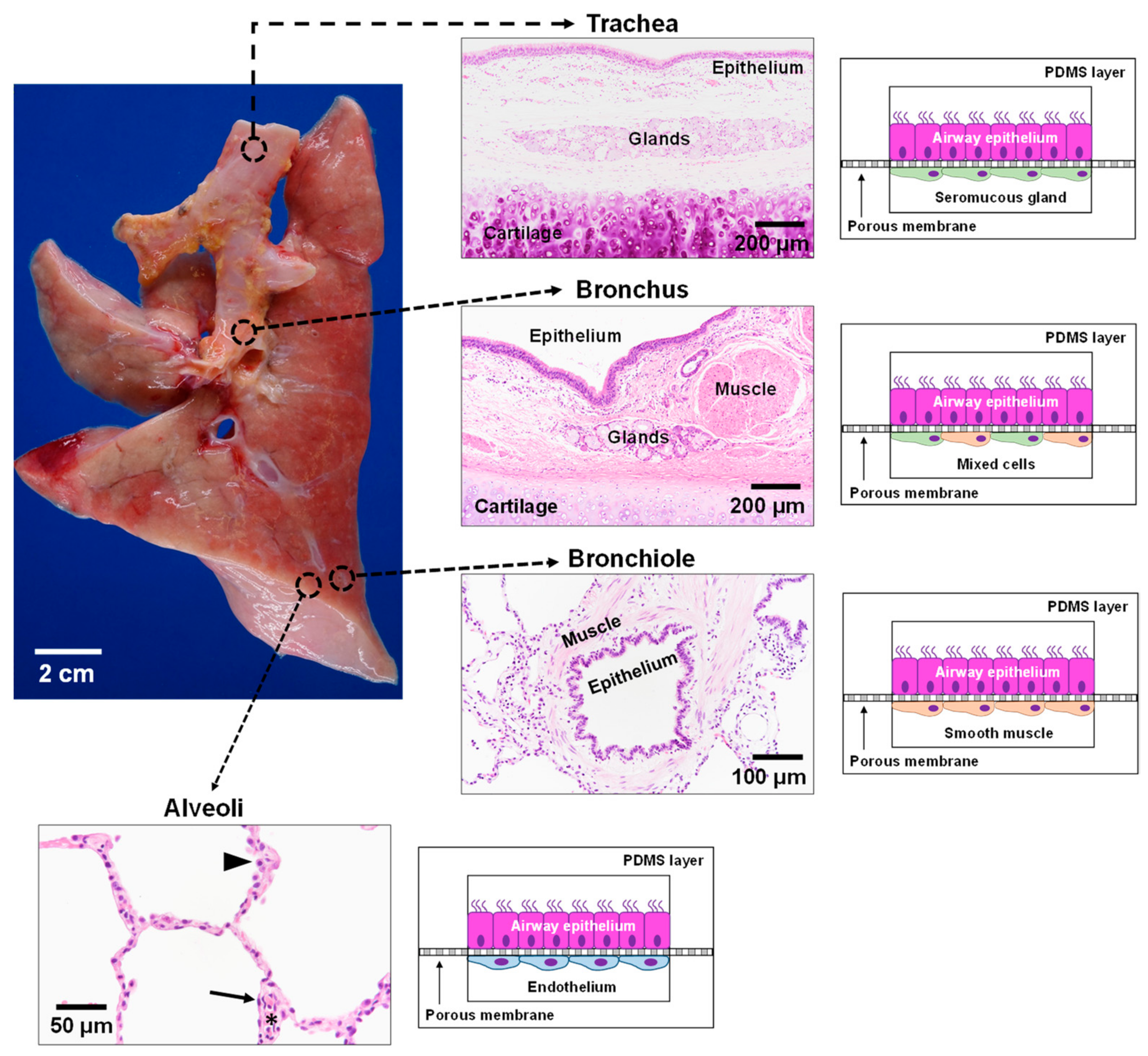
3.3. Lung
4. Clinical Trials and N-of-1 Studies
5. Animal Models in CF
6. Organs-on-a-Chip
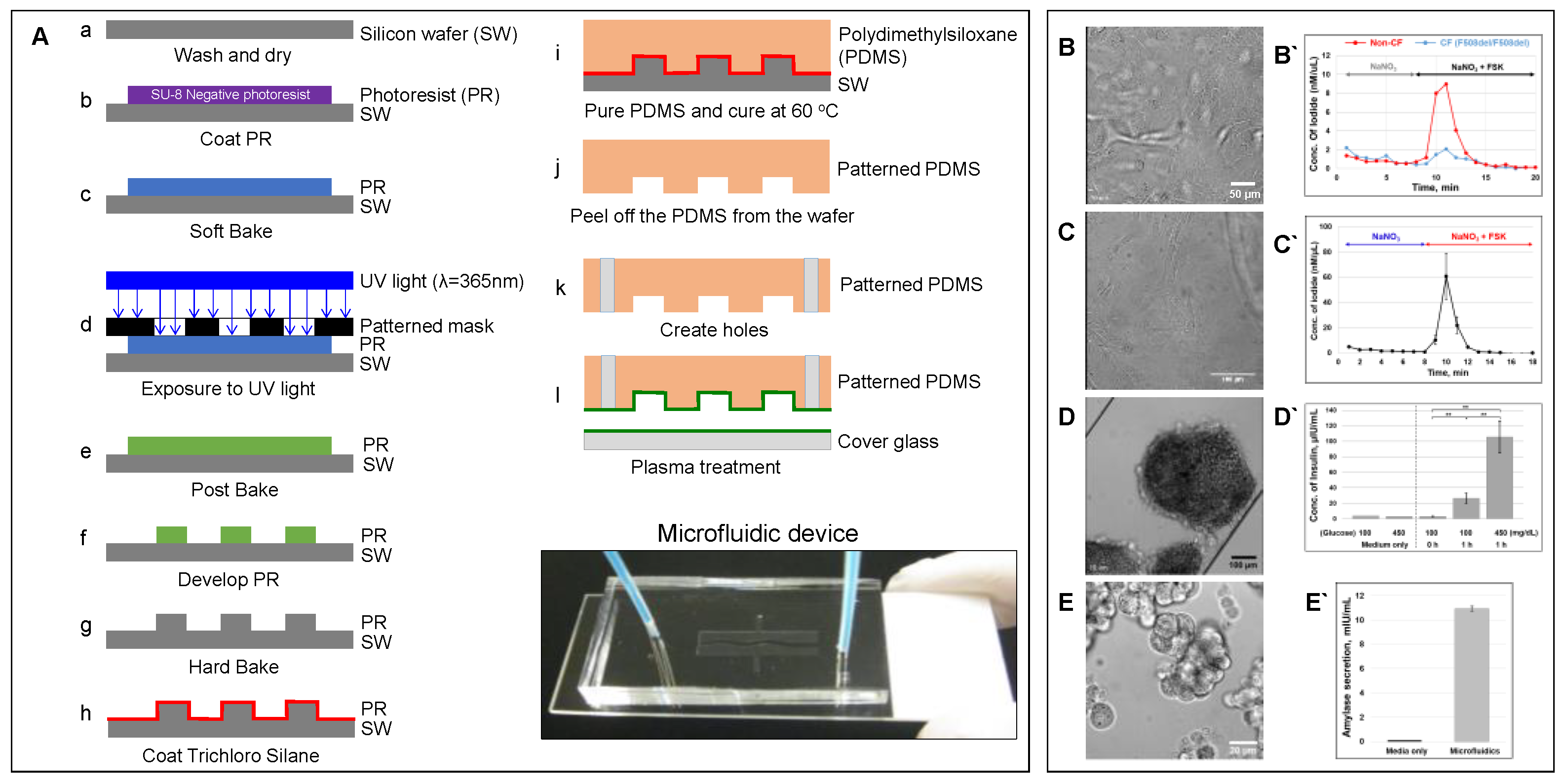
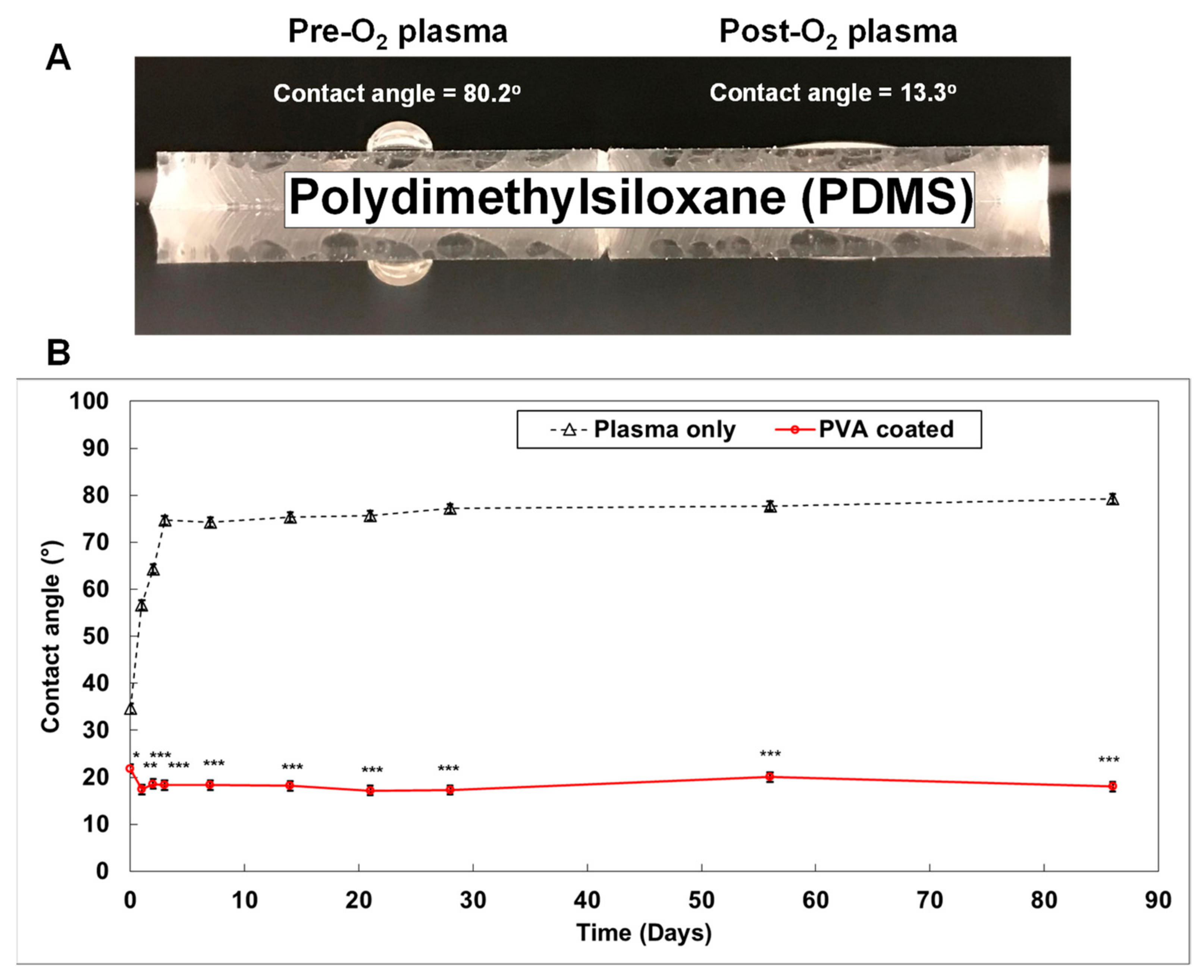
6.1. Microfluidic-Based Organs-on-a-Chip
6.2. Advantages of Organs-on-a-Chip
6.3. Limitations of Organs-on-a-Chip
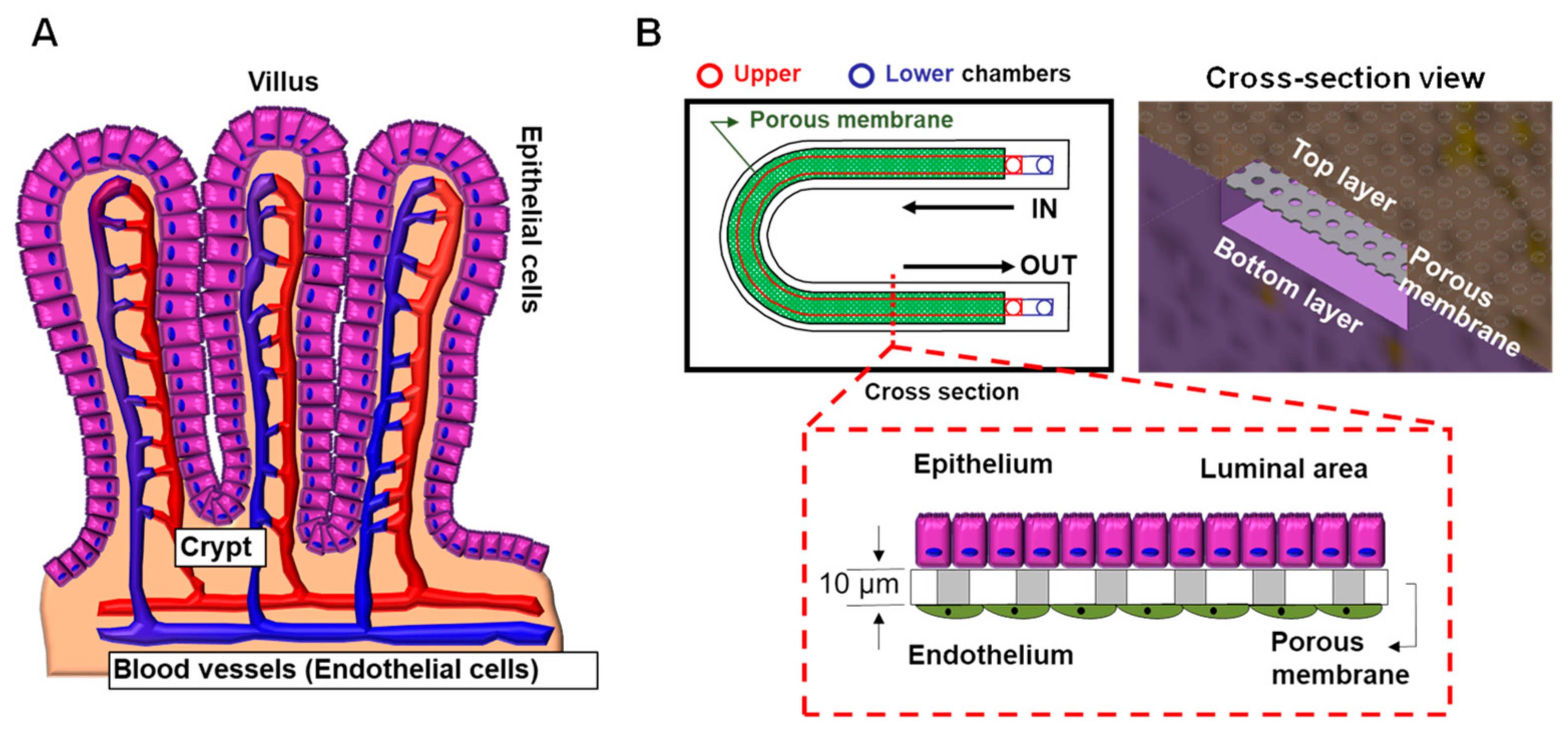
6.4. CF Modeling in the Lung
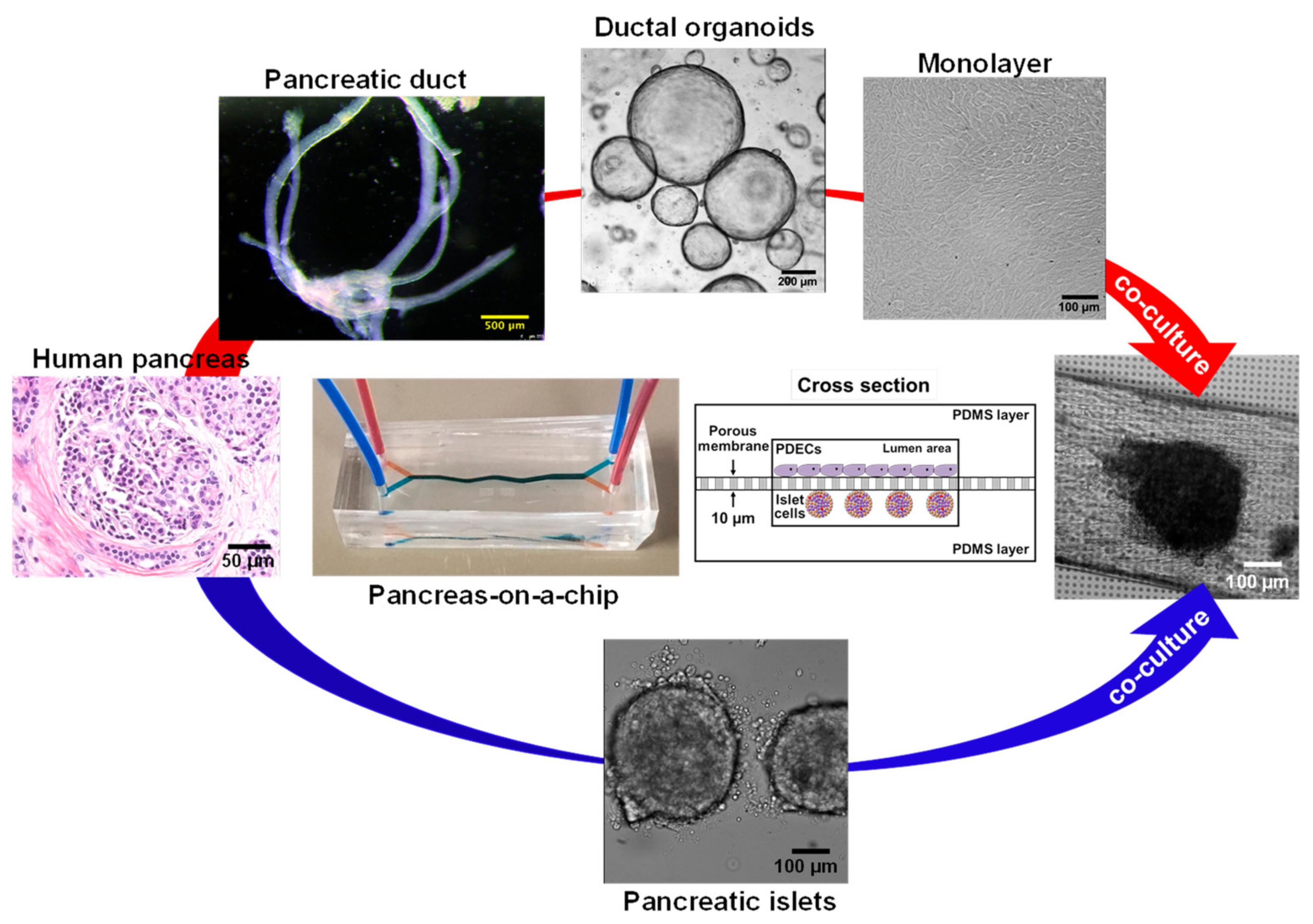
6.5. CF modeling in the Pancreas
6.6. CF Modeling in the GI Tract
6.7. Precision Medicine Using Organs-on-a-Chip
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathologic study. Am. J. Dis. Child. 1938, 56, 344–399. [Google Scholar] [CrossRef]
- Fanen, P.; Wohlhuter-Haddad, A.; Hinzpeter, A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int. J. Biochem. Cell Biol. 2014, 52, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Harutyunyan, M.; Huang, Y.J.; Mun, K.S.; Yang, F.M.Y.; Arora, K.; Naren, A.P. Personalized medicine in CF: From modulator development to therapy for cystic fibrosis patients with rare CFTR mutations. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, 1529–1543. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Doring, G. Cystic fibrosis. Lancet 2003, 361, 681–689. [Google Scholar] [CrossRef]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- Cebotaru, L.; Rapino, D.; Cebotaru, V.; Guggino, W.B. Correcting the cystic fibrosis disease mutant, A455E CFTR. PLoS ONE 2014, 9, e85183. [Google Scholar] [CrossRef] [PubMed]
- Dugueperoux, I.; De Braekeleer, M. The CFTR 3849+10kbC->T and 2789+5G->A alleles are associated with a mild CF phenotype. Eur. Respir. J. 2005, 25, 468–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, S.; Penque, D.; Garcia, S.; Gomes, A.; Farinha, C.; Mata, L.; Gulbenkian, S.; Gil-Ferreira, K.; Duarte, A.; Pacheco, P.; et al. Cystic fibrosis patients with the 3272-26A-->G mutation have mild disease, leaky alternative mRNA splicing, and CFTR protein at the cell membrane. Hum. Mutat. 1999, 14, 133–144. [Google Scholar] [CrossRef]
- Yeh, J.T.; Yu, Y.C.; Hwang, T.C. Structural mechanisms for defective CFTR gating caused by the Q1412X mutation, a severe Class VI pathogenic mutation in cystic fibrosis. J. Physiol. 2019, 597, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Tata, F.; Stanier, P.; Wicking, C.; Halford, S.; Kruyer, H.; Lench, N.J.; Scambler, P.J.; Hansen, C.; Braman, J.C.; Williamson, R.; et al. Cloning the mouse homolog of the human cystic fibrosis transmembrane conductance regulator gene. Genomics 1991, 10, 301–307. [Google Scholar] [CrossRef]
- Colledge, W.H.; Abella, B.S.; Southern, K.W.; Ratcliff, R.; Jiang, C.; Cheng, S.H.; MacVinish, L.J.; Anderson, J.R.; Cuthbert, A.W.; Evans, M.J. Generation and characterization of a delta F508 cystic fibrosis mouse model. Nat. Genet. 1995, 10, 445–452. [Google Scholar] [CrossRef]
- Zhou, L.; Dey, C.R.; Wert, S.E.; DuVall, M.D.; Frizzell, R.A.; Whitsett, J.A. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science 1994, 266, 1705–1708. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sui, H.; Fisher, J.T.; Yan, Z.; Liu, X.; Cho, H.J.; Joo, N.S.; Zhang, Y.; Zhou, W.; Yi, Y.; et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Investig. 2010, 120, 3149–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, S.L.; Irimura, T.; Landers, R.A.; Bridges, C.D. The carbohydrate of bovine interstitial retinol-binding protein. Prog. Clin. Biol. Res. 1985, 190, 111–128. [Google Scholar] [PubMed]
- Ostedgaard, L.S.; Rogers, C.S.; Dong, Q.; Randak, C.O.; Vermeer, D.W.; Rokhlina, T.; Karp, P.H.; Welsh, M.J. Processing and function of CFTR-DeltaF508 are species-dependent. Proc. Natl. Acad. Sci. USA 2007, 104, 15370–15375. [Google Scholar] [CrossRef] [Green Version]
- Trezise, A.E.; Szpirer, C.; Buchwald, M. Localization of the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) in the rat to chromosome 4 and implications for the evolution of mammalian chromosomes. Genomics 1992, 14, 869–874. [Google Scholar] [CrossRef]
- Dupuit, F.; Bout, A.; Hinnrasky, J.; Fuchey, C.; Zahm, J.M.; Imler, J.L.; Pavirani, A.; Valerio, D.; Puchelle, E. Expression and localization of CFTR in the rhesus monkey surface airway epithelium. Gene Ther. 1995, 2, 156–163. [Google Scholar] [PubMed]
- Harris, A. Towards an ovine model of cystic fibrosis. Hum. Mol. Genet. 1997, 6, 2191–2194. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Perisse, I.V.; Cotton, C.U.; Regouski, M.; Meng, Q.; Domb, C.; Van Wettere, A.J.; Wang, Z.; Harris, A.; White, K.L.; et al. A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. JCI Insight 2018, 3, e123529. [Google Scholar] [CrossRef]
- McCarron, A.; Donnelley, M.; Parsons, D. Airway disease phenotypes in animal models of cystic fibrosis. Respir. Res. 2018, 19, 54. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Quon, B.S.; Rowe, S.M. New and emerging targeted therapies for cystic fibrosis. BMJ 2016, 352, i859. [Google Scholar] [CrossRef] [Green Version]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2010, 363, 1991–2003. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Eckford, P.D.; Li, C.; Ramjeesingh, M.; Bear, C.E. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 2012, 287, 36639–36649. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Lukacs, G.L. Fixing cystic fibrosis by correcting CFTR domain assembly. J. Cell. Biol. 2012, 199, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Burton, B.; Huang, C.J.; Worley, J.; Cao, D.; Johnson, J.P., Jr.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012, 11, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Flume, P.A.; Liou, T.G.; Borowitz, D.S.; Li, H.; Yen, K.; Ordonez, C.L.; Geller, D.E.; Group, V.X.S. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012, 142, 718–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuk, K.; Taylor-Cousar, J.L. Lumacaftor and ivacaftor in the management of patients with cystic fibrosis: Current evidence and future prospects. Ther. Adv. Respir. Dis. 2015, 9, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Tezacaftor/ivacaftor for cystic fibrosis. Aust. Prescr. 2019, 42, 174–175. [CrossRef]
- Hoy, S.M. Elexacaftor/Ivacaftor/Tezacaftor: First Approval. Drugs 2019, 79, 2001–2007. [Google Scholar] [CrossRef]
- Bear, C.E. A Therapy for Most with Cystic Fibrosis. Cell 2020, 180, 211. [Google Scholar] [CrossRef]
- Tindell, W.; Su, A.; Oros, S.M.; Rayapati, A.O.; Rakesh, G. Trikafta and Psychopathology in Cystic Fibrosis: A Case Report. Psychosomatics 2020, 61, 735–738. [Google Scholar] [CrossRef]
- Elexacaftor/tezacaftor/ivacaftor (Trikafta) for cystic fibrosis. Med. Lett. Drugs Ther. 2020, 62, 5–7.
- Whitcomb, D.C.; Lowe, M.E. Human pancreatic digestive enzymes. Dig. Dis. Sci. 2007, 52, 1–17. [Google Scholar] [CrossRef]
- Logsdon, C.D.; Ji, B. The role of protein synthesis and digestive enzymes in acinar cell injury. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Wilschanski, M.; Novak, I. The cystic fibrosis of exocrine pancreas. Cold Spring Harb. Perspect. Med. 2013, 3, a009746. [Google Scholar] [CrossRef]
- Ahmed, N.; Corey, M.; Forstner, G.; Zielenski, J.; Tsui, L.C.; Ellis, L.; Tullis, E.; Durie, P. Molecular consequences of cystic fibrosis transmembrane regulator (CFTR) gene mutations in the exocrine pancreas. Gut 2003, 52, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.K.; Haupt, M.E.; Geller, D.E.; Hall, J.A.; Quintana Diez, P.M. Less common etiologies of exocrine pancreatic insufficiency. World J. Gastroenterol. 2017, 23, 7059–7076. [Google Scholar] [CrossRef]
- Da Silva Xavier, G. The Cells of the Islets of Langerhans. J. Clin. Med. 2018, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionescu-Tirgoviste, C.; Gagniuc, P.A.; Gubceac, E.; Mardare, L.; Popescu, I.; Dima, S.; Militaru, M. A 3D map of the islet routes throughout the healthy human pancreas. Sci. Rep. 2015, 5, 14634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brissova, M.; Fowler, M.J.; Nicholson, W.E.; Chu, A.; Hirshberg, B.; Harlan, D.M.; Powers, A.C. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J. Histochem. Cytochem. 2005, 53, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Orci, L.; Baetens, D.; Ravazzola, M.; Stefan, Y.; Malaisse-Lagae, F. Pancreatic polypeptide and glucagon: Non-random distribution in pancreatic islets. Life Sci. 1976, 19, 1811–1815. [Google Scholar] [CrossRef]
- Ichii, H.; Inverardi, L.; Pileggi, A.; Molano, R.D.; Cabrera, O.; Caicedo, A.; Messinger, S.; Kuroda, Y.; Berggren, P.O.; Ricordi, C. A novel method for the assessment of cellular composition and beta-cell viability in human islet preparations. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2005, 5, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Kayani, K.; Mohammed, R.; Mohiaddin, H. Cystic Fibrosis-Related Diabetes. Front. Endocrinol. 2018, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelsey, R.; Koivula, F.N.M.; McClenaghan, N.H.; Kelly, C. Cystic Fibrosis-Related Diabetes: Pathophysiology and Therapeutic Challenges. Clin. Med. Insights Endocrinol. Diabetes 2019, 12, 1179551419851770. [Google Scholar] [CrossRef] [Green Version]
- Melamed, P.; Melamed, F. Chronic metabolic acidosis destroys pancreas. JOP J. Pancreas 2014, 15, 552–560. [Google Scholar]
- Mun, K.S.; Arora, K.; Huang, Y.; Yang, F.; Yarlagadda, S.; Ramananda, Y.; Abu-El-Haija, M.; Palermo, J.J.; Appakalai, B.N.; Nathan, J.D.; et al. Patient-derived pancreas-on-a-chip to model cystic fibrosis-related disorders. Nat. Commun. 2019, 10, 3124. [Google Scholar]
- Ashammakhi, N.; Nasiri, R.; Barros, N.R.; Tebon, P.; Thakor, J.; Goudie, M.; Shamloo, A.; Martin, M.G.; Khademhosseini, A. Gut-on-a-chip: Current progress and future opportunities. Biomaterials 2020, 255, 120196. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Dorsey, J.; Gonska, T. Bacterial overgrowth, dysbiosis, inflammation, and dysmotility in the Cystic Fibrosis intestine. J. Cyst. Fibros. 2017, 16 (Suppl. 2), S14–S23. [Google Scholar] [CrossRef] [Green Version]
- Dray, X.; Bienvenu, T.; Desmazes-Dufeu, N.; Dusser, D.; Marteau, P.; Hubert, D. Distal intestinal obstruction syndrome in adults with cystic fibrosis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2004, 2, 498–503. [Google Scholar] [CrossRef]
- Van der Doef, H.P.; Kokke, F.T.; van der Ent, C.K.; Houwen, R.H. Intestinal obstruction syndromes in cystic fibrosis: Meconium ileus, distal intestinal obstruction syndrome, and constipation. Curr. Gastroenterol. Rep. 2011, 13, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: Pathophysiology and therapeutic targets. Thorax 2016, 71, 284–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.L.; Saint-Criq, V.; Hwang, T.C.; Csanady, L. Ion channels as targets to treat cystic fibrosis lung disease. J. Cyst. Fibros. 2018, 17, S22–S27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amrani, Y.; Panettieri, R.A. Airway smooth muscle: Contraction and beyond. Int. J. Biochem. Cell Biol. 2003, 35, 272–276. [Google Scholar] [CrossRef]
- Comhair, S.A.; Xu, W.; Mavrakis, L.; Aldred, M.A.; Asosingh, K.; Erzurum, S.C. Human primary lung endothelial cells in culture. Am. J. Respir. Cell Mol. Biol. 2012, 46, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreda, S.M.; Mall, M.; Mengos, A.; Rochelle, L.; Yankaskas, J.; Riordan, J.R.; Boucher, R.C. Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell. 2005, 16, 2154–2167. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, J.F.; Yankaskas, J.R.; Ernst, S.A.; Yang, Y.; Marino, C.R.; Boucher, R.C.; Cohn, J.A.; Wilson, J.M. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat. Genet. 1992, 2, 240–248. [Google Scholar] [CrossRef]
- Regnier, A.; Dannhoffer, L.; Blouquit-Laye, S.; Bakari, M.; Naline, E.; Chinet, T. Expression of cystic fibrosis transmembrane conductance regulator in the human distal lung. Hum. Pathol. 2008, 39, 368–376. [Google Scholar] [CrossRef]
- Balfour-Lynn, I.M.; Elborn, J.S. “CF asthma”: What is it and what do we do about it? Thorax 2002, 57, 742–748. [Google Scholar] [CrossRef] [Green Version]
- Hays, S.R.; Ferrando, R.E.; Carter, R.; Wong, H.H.; Woodruff, P.G. Structural changes to airway smooth muscle in cystic fibrosis. Thorax 2005, 60, 226–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awadalla, M.; Miyawaki, S.; Abou Alaiwa, M.H.; Adam, R.J.; Bouzek, D.C.; Michalski, A.S.; Fuld, M.K.; Reynolds, K.J.; Hoffman, E.A.; Lin, C.L.; et al. Early airway structural changes in cystic fibrosis pigs as a determinant of particle distribution and deposition. Ann. Biomed. Eng. 2014, 42, 915–927. [Google Scholar] [CrossRef] [Green Version]
- Meyerholz, D.K.; Stoltz, D.A.; Namati, E.; Ramachandran, S.; Pezzulo, A.A.; Smith, A.R.; Rector, M.V.; Suter, M.J.; Kao, S.; McLennan, G.; et al. Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am. J. Respir. Crit. Care Med. 2010, 182, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Michoud, M.C.; Robert, R.; Hassan, M.; Moynihan, B.; Haston, C.; Govindaraju, V.; Ferraro, P.; Hanrahan, J.W.; Martin, J.G. Role of the cystic fibrosis transmembrane conductance channel in human airway smooth muscle. Am. J. Respir. Cell. Mol. Biol. 2009, 40, 217–222. [Google Scholar] [CrossRef]
- Robert, R.; Norez, C.; Becq, F. Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl-transport of mouse aortic smooth muscle cells. J. Physiol. 2005, 568, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Vandebrouck, C.; Melin, P.; Norez, C.; Robert, R.; Guibert, C.; Mettey, Y.; Becq, F. Evidence that CFTR is expressed in rat tracheal smooth muscle cells and contributes to bronchodilation. Respir. Res. 2006, 7, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavelle, G.M.; White, M.M.; Browne, N.; McElvaney, N.G.; Reeves, E.P. Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences. Biomed. Res. Int. 2016, 2016, 5258727. [Google Scholar] [CrossRef] [Green Version]
- Norez, C.; Jayle, C.; Becq, F.; Vandebrouck, C. Bronchorelaxation of the human bronchi by CFTR activators. Pulm. Pharmacol. Ther. 2014, 27, 38–43. [Google Scholar] [CrossRef]
- Cook, D.P.; Rector, M.V.; Bouzek, D.C.; Michalski, A.S.; Gansemer, N.D.; Reznikov, L.R.; Li, X.; Stroik, M.R.; Ostedgaard, L.S.; Alaiwa, M.H.A.; et al. Cystic Fibrosis Transmembrane Conductance Regulator in Sarcoplasmic Reticulum of Airway Smooth Muscle. Implications for Airway Contractility. Am. J. Respir. Crit. Care Med. 2016, 193, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Clancy, J.P. Cystic Fibrosis Transmembrane Conductance Regulator Function in Airway Smooth Muscle. A Novel Role in Cystic Fibrosis Airway Obstruction. Am. J. Respir. Crit. Care Med. 2016, 193, 352–353. [Google Scholar] [CrossRef]
- Döring, G.; Elborn, J.S.; Johannesson, M.; de Jonge, H.; Griese, M.; Smyth, A.; Heijerman, H. Clinical trials in cystic fibrosis. J. Cyst. Fibros. 2007, 6, 85–99. [Google Scholar] [CrossRef] [Green Version]
- Sucharew, H.; Goss, C.H.; Millard, S.P.; Ramsey, B.W.; Cystic Fibrosis Therapeutics Development Network. Respiratory adverse event profiles in cystic fibrosis placebo subjects in short- and long-term inhaled therapy trials. Contemp. Clin. Trials 2006, 27, 561–570. [Google Scholar] [CrossRef]
- VanDevanter, D.R.; Mayer-Hamblett, N. Innovating cystic fibrosis clinical trial designs in an era of successful standard of care therapies. Curr. Opin. Pulm. Med. 2017, 23, 530–535. [Google Scholar] [CrossRef] [PubMed]
- VanDevanter, D.R.; Konstan, M.W. Outcome measures for clinical trials assessing treatment of cystic fibrosis lung disease. Clin. Investig. 2012, 2, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griesenbach, U.; Alton, E.W. Recent advances in understanding and managing cystic fibrosis transmembrane conductance regulator dysfunction. F1000Prime Rep. 2015, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- McGarry, M.E.; Illek, B.; Ly, N.P.; Zlock, L.; Olshansky, S.; Moreno, C.; Finkbeiner, W.E.; Nielson, D.W. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr. Pulmonol. 2017, 52, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Lillie, E.O.; Patay, B.; Diamant, J.; Issell, B.; Topol, E.J.; Schork, N.J. The n-of-1 clinical trial: The ultimate strategy for individualizing medicine? Pers. Med. 2011, 8, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Kerem, E. Cystic fibrosis: Priorities and progress for future therapies. Paediatr. Respir. Rev. 2017, 24, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Kim, H.J.; Fraser, J.P.; Shea, D.E.; Khan, M.; Bahinski, A.; Hamilton, G.A.; Ingber, D.E. Microfabrication of human organs-on-chips. Nat. Protoc. 2013, 8, 2135–2157. [Google Scholar] [CrossRef]
- Knight, A. Systematic reviews of animal experiments demonstrate poor contributions toward human healthcare. Rev. Recent Clin. Trials 2008, 3, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konar, D.; Devarasetty, M.; Yildiz, D.V.; Atala, A.; Murphy, S.V. Lung-On-A-Chip Technologies for Disease Modeling and Drug Development. Biomed. Eng. Comput. Biol. 2016, 7, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.J.; Spence, J.R. In Vitro Models to Study Human Lung Development, Disease and Homeostasis. Physiology 2017, 32, 246–260. [Google Scholar] [CrossRef]
- Gökaltun, A.; Kang, Y.B.; Yarmush, M.L.; Usta, O.B.; Asatekin, A. Simple Surface Modification of Poly(dimethylsiloxane) via Surface Segregating Smart Polymers for Biomicrofluidics. Sci. Rep. 2019, 9, 7377. [Google Scholar] [CrossRef] [Green Version]
- Firpo, G.; Angeli, E.; Repetto, L.; Valbusa, U. Permeability thickness dependence of polydimethylsiloxane (PDMS) membranes. J. Membr. Sci. 2015, 481, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Trantidou, T.; Elani, Y.; Parsons, E.; Ces, O. Hydrophilic surface modification of PDMS for droplet microfluidics using a simple, quick, and robust method via PVA deposition. Microsyst. Nanoeng. 2017, 3, 16091. [Google Scholar] [CrossRef]
- Mukhopadhyay, R. When PDMS isn’t the best. What are its weaknesses, and which other polymers can researchers add to their toolboxes? Anal. Chem. 2007, 79, 3248–3253. [Google Scholar] [CrossRef]
- Shirure, V.S.; George, S.C. Design considerations to minimize the impact of drug absorption in polymer-based organ-on-a-chip platforms. Lab Chip 2017, 17, 681–690. [Google Scholar] [CrossRef]
- Akther, F.; Yakob, S.B.; Nguyen, N.T.; Ta, H.T. Surface Modification Techniques for Endothelial Cell Seeding in PDMS Microfluidic Devices. Biosensors 2020, 10, 182. [Google Scholar] [CrossRef]
- Benam, K.H.; Villenave, R.; Lucchesi, C.; Varone, A.; Hubeau, C.; Lee, H.H.; Alves, S.E.; Salmon, M.; Ferrante, T.C.; Weaver, J.C.; et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat. Methods 2016, 13, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Brand-Saberi, B.E.M.; Schafer, T. Trachea: Anatomy and physiology. Thorac. Surg. Clin. 2014, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Breeze, R.; Turk, M. Cellular structure, function and organization in the lower respiratory tract. Environ. Health Perspect. 1984, 55, 3–24. [Google Scholar] [CrossRef]
- Nunn, J.F. Chapter 2—Functional anatomy of the respiratory tract. In Nunn’s Applied Respiratory Physiology, 4th ed.; Nunn, J.F., Ed.; Butterworth-Heinemann: Oxford, UK, 1993; pp. 13–35. [Google Scholar] [CrossRef]
- Mescher, A. Junqueira’s Basic Histology Text & Atlas, 14th ed.; McGraw-Hill Medical: New York, PA, USA, 2016. [Google Scholar]
- Finkbeiner, W.E.; Zlock, L.T.; Mehdi, I.; Widdicombe, J.H. Cultures of human tracheal gland cells of mucous or serous phenotype. In Vitro Cell. Dev. Biol Anim. 2010, 46, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Jun, Y.; Lee, J.; Choi, S.; Yang, J.H.; Sander, M.; Chung, S.; Lee, S.H. In vivo-mimicking microfluidic perfusion culture of pancreatic islet spheroids. Sci. Adv. 2019, 5, eaax4520. [Google Scholar] [CrossRef] [Green Version]
- Glieberman, A.L.; Pope, B.D.; Zimmerman, J.F.; Liu, Q.; Ferrier, J.P.; Kenty, J.H.R.; Schrell, A.M.; Mukhitov, N.; Shores, K.L.; Tepole, A.B.; et al. Synchronized stimulation and continuous insulin sensing in a microfluidic human Islet on a Chip designed for scalable manufacturing. Lab Chip 2019, 19, 2993–3010. [Google Scholar] [CrossRef] [PubMed]
- Zbinden, A.; Marzi, J.; Schlunder, K.; Probst, C.; Urbanczyk, M.; Black, S.; Brauchle, E.M.; Layland, S.L.; Kraushaar, U.; Duffy, G.; et al. Non-invasive marker-independent high content analysis of a microphysiological human pancreas-on-a-chip model. Matrix Biol. 2020, 85, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Wang, Y.; Chen, W.; Li, Z.; Su, W.; Guo, Y.; Deng, P.; Qin, J. Engineering human islet organoids from iPSCs using an organ-on-chip platform. Lab Chip 2019, 19, 948–958. [Google Scholar] [CrossRef]
- Sontheimer-Phelps, A.; Chou, D.B.; Tovaglieri, A.; Ferrante, T.C.; Duckworth, T.; Fadel, C.; Frismantas, V.; Sutherland, A.D.; Jalili-Firoozinezhad, S.; Kasendra, M.; et al. Human Colon-on-a-Chip Enables Continuous In Vitro Analysis of Colon Mucus Layer Accumulation and Physiology. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 507–526. [Google Scholar] [CrossRef] [Green Version]
- Choe, A.; Ha, S.K.; Choi, I.; Choi, N.; Sung, J.H. Microfluidic Gut-liver chip for reproducing the first pass metabolism. Biomed. Microdevices 2017, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, V.; Telesco, M.; Corrado, B.; Rosiello, V.; Urciuolo, F.; Netti, P.A.; Imparato, G. Intestine-Liver Axis On-Chip Reveals the Intestinal Protective Role on Hepatic Damage by Emulating Ethanol First-Pass Metabolism. Front. Bioeng. Biotechnol. 2020, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 2016, 21, 1399–1411. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogden, H.L.; Kim, H.; Wikenheiser-Brokamp, K.A.; Naren, A.P.; Mun, K.S. Cystic Fibrosis Human Organs-on-a-Chip. Micromachines 2021, 12, 747. https://doi.org/10.3390/mi12070747
Ogden HL, Kim H, Wikenheiser-Brokamp KA, Naren AP, Mun KS. Cystic Fibrosis Human Organs-on-a-Chip. Micromachines. 2021; 12(7):747. https://doi.org/10.3390/mi12070747
Chicago/Turabian StyleOgden, Herbert Luke, Hoyeol Kim, Kathryn A. Wikenheiser-Brokamp, Anjaparavanda P. Naren, and Kyu Shik Mun. 2021. "Cystic Fibrosis Human Organs-on-a-Chip" Micromachines 12, no. 7: 747. https://doi.org/10.3390/mi12070747
APA StyleOgden, H. L., Kim, H., Wikenheiser-Brokamp, K. A., Naren, A. P., & Mun, K. S. (2021). Cystic Fibrosis Human Organs-on-a-Chip. Micromachines, 12(7), 747. https://doi.org/10.3390/mi12070747