1. Introduction
Organoids are in vitro mini organs that mimic some of the functions of in vivo tissues or organs and are highly expected to contribute to applications such as drug discovery, regenerative medicine, and as models to study developmental biology [
1,
2,
3]. Nevertheless, one of the bottlenecks of current organoid culture methods that could be hindering further progress in the field is the lack of control of the shape of organoids. In most cases, organoids are cultured simply as a cluster of cells, either free-floating or in an extracellular matrix (ECM) hydrogel, and allowed to self-organize with only molecular cues for guidance. However, this oversimplified method of culturing organoids does not take into account the physical boundaries and the local environment surrounding the cells that contribute important cues for the formation and function of organoids, such as mechanical stress distribution in tissues and ECM composition [
4,
5]. Several methods have been employed to enhance the control of organoid shape, including micromanufacturing hydrogels with pre-defined shapes [
6,
7], laser-ablation of microchannels in hydrogels [
8], and utilizing microfilaments as scaffolds for cells to attach onto [
9,
10]. Still, the patterns and shapes that can be achieved by these technologies are mainly limited to simple linear geometries only.
On the other hand, advances in 3D-printing technologies have greatly increased the complexity and sophistication of configurations in which cells can be formed into organoids [
11,
12]. Sacrificial templating, in which a sacrificial material is used to provide temporary structural support during the fabrication process, has been widely applied to create complex 3D tissues. For example, the freeform reversible embedding of suspended hydrogels (FRESH) method was used with a gelatin support material to fabricate a heart ventricle using collagen and cell-laden bio-inks [
13], and water-soluble Pluronic or carbohydrate glass inks have been used as sacrificial molds to create vascular channels in tissue constructs [
14,
15]. However, due to the relatively large sizes of the fabricated 3D tissues or organoids, imaging resolution in the
z-direction tends to be poor. Cryo- or paraffin-sectioning methods are commonly used to visualize deeper sections of large tissue samples, but not for obtaining z information, as it is difficult to reconstitute a whole image of the sample from the sliced sections. On the other hand, high-resolution images can be obtained with higher magnification lenses, but the focal depths of these objective lenses are only around tens to hundreds of micrometers, which is inadequate for samples that are of millimeter order in size. Inversely, low magnification lenses have larger focal depths but with compromised resolution. To overcome this issue, clearing reagents are often used to render samples transparent to enable deeper scanning of the sample with a laser, but the focal depth issue remains when a low magnification lens is used.
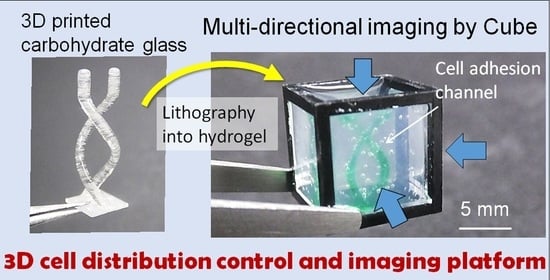
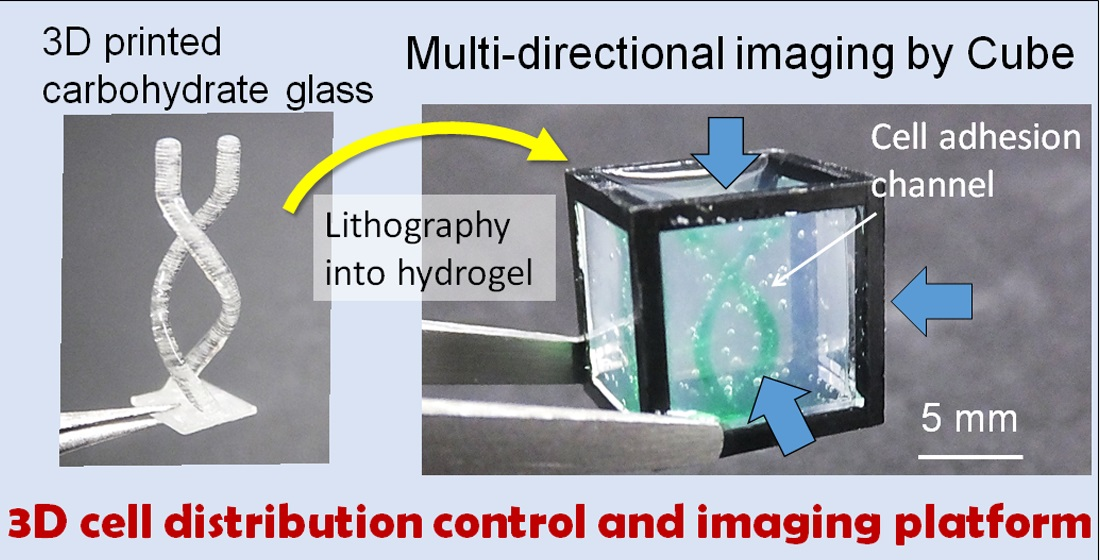
We previously developed a gel cube device made of a polycarbonate frame, outer agarose gel walls, and an inner ECM hydrogel in which tissue samples are supported [
16]. We showed that this cube device can be used to control initial cell seeding geometry with simple shapes such as cylinders or prisms for highly repeatable experiments [
6] and to obtain high-resolution imaging on a large-scale by acquiring and merging images of the sample from all six sides of the cube [
17]. In this paper, to develop a platform that simultaneously allows cell seeding control and high-resolution imaging of large-scale samples for organoid culture, we utilized 3D-printed sacrificial molds to generate more complex patterns in the hydrogel in the cube device compared with the limited simple patterns obtainable by photocurable resin molds in our previous work. We opted to use carbohydrate glass originally developed by Miller et al. [
15,
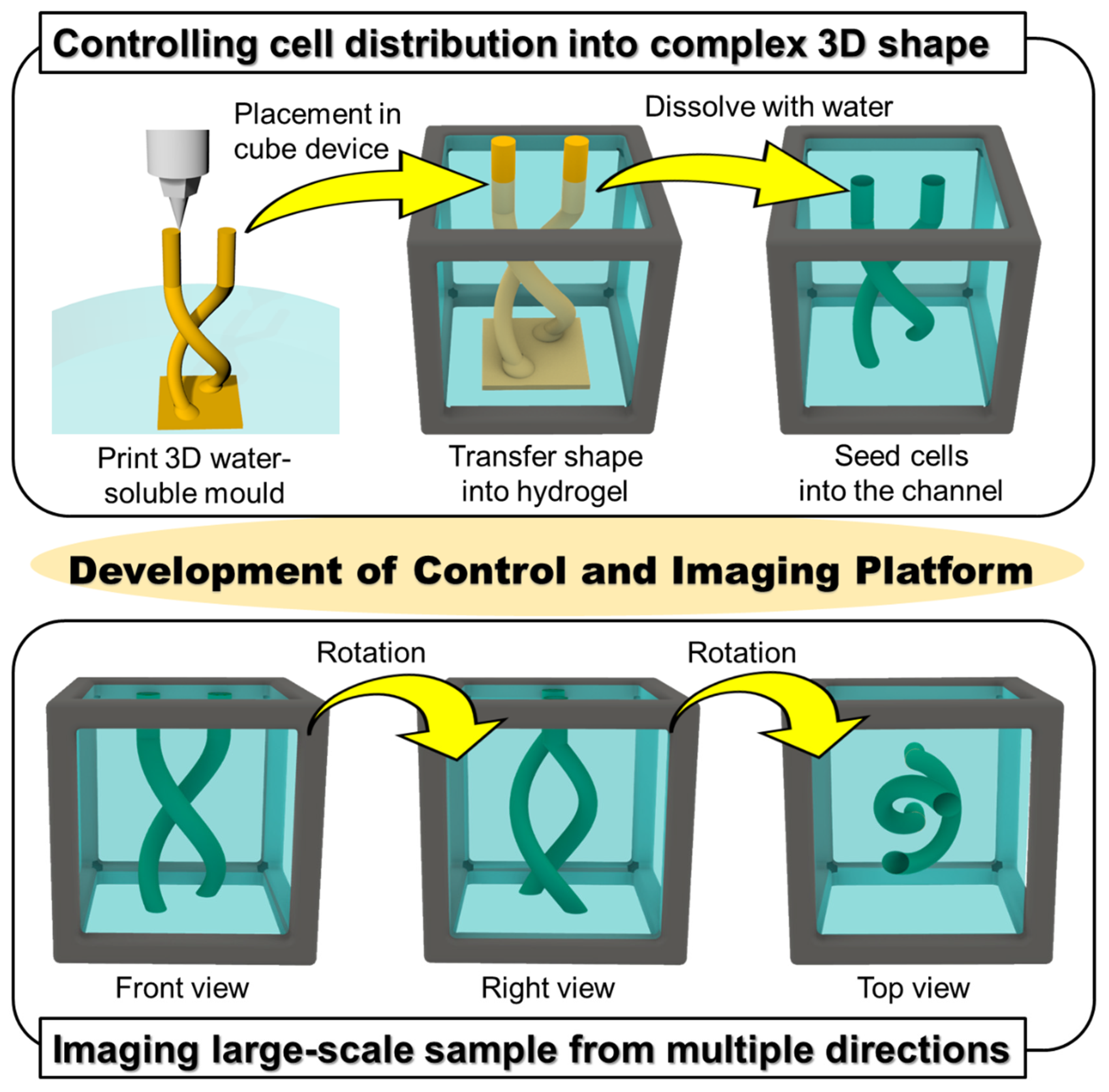
18], which is now commercially available, as the non-cytotoxic resin for our molds. To make complex shapes in the cube device, printed molds of the desired design were placed in the hydrogel in the cube device, and once the gel cures, the mold can be removed simply by immersing the cube in an aqueous solution, leaving only a patterned shape in the hydrogel (
Figure 1). We also designed and fabricated accessories using a stereolithography 3D printer to align the position of the mold in the cube, which facilitates the precise transfer of the mold shape to the hydrogel. We first optimized the printing parameters that would enable us to accurately print the sacrificial mold structures, then compared the printed mold dimensions with that of the pattern formed in the hydrogel to validate the preservation of structural accuracy after transfer of the mold shape to the hydrogel. Finally, to demonstrate the advantages of integrating sacrificial templating with the gel cube device, we seeded endothelial cells in the formed shape, and performed multi-directional imaging to visualize the tissue construct.
2. Materials and Methods
2.1. Materials
Polydimethylsiloxane (PDMS; Silpot 184, Dow Corning, Midland, MI, USA) was used to make the PDMS sidewalls of the gel cube device and flexible silicone printing surfaces. Water-soluble carbohydrate glass (CG3357, Volumetric, Houston, TX, USA) was used for mold printing. Poly (D, L-lactide-co-glycolide) (PLGA; 26269-10, Polysciences, Warrington, PA, USA) and chloroform (08402-55, Nacalai Tesque, Kyoto, Japan) were used to prepare PLGA solution as a hydrophobic coating for the mold. The compound 2-propanol (29112-63, FUJIFILM Wako Pure Chemical, Osaka, Japapn) was used for cleaning the cube devices and to remove excess PLGA coating. An amount of 3 mg/mL collagen type I derived from porcine tendon (Cellmatrix type IA, Nitta Gelatin, Morrisville, NC, USA) was used as a hydrogel. Phosphate-buffered saline (PBS; 14190-144, Gibco, Waltham, MA, USA) was used for the dissolution of water-soluble molds, retention of hydrogel, and in cell culture. Human umbilical vein endothelial cells (HUVEC; C2519A, Lonza, Basel, Switzerland), EGM-2 medium (CC-3162, Lonza), penicillin–streptomycin (Pen-strep; 15140-122, Gibco), Trypsin-EDTA (25200-056, Gibco), and Trypsin neutralizing solution (TNS; HK-3220, Kurabo, Osaka, Japan) were used in cell culture. Alexa fluor 488 Phalloidin (A12379, Thermo Fisher Scientific, Waltham, MA, USA) was used for immunofluorescence staining. Fluorescent microbeads with 45 µm diameter (18242-2, Polysciences, Warrington, PA, USA) were used for measurements of the gel channels.
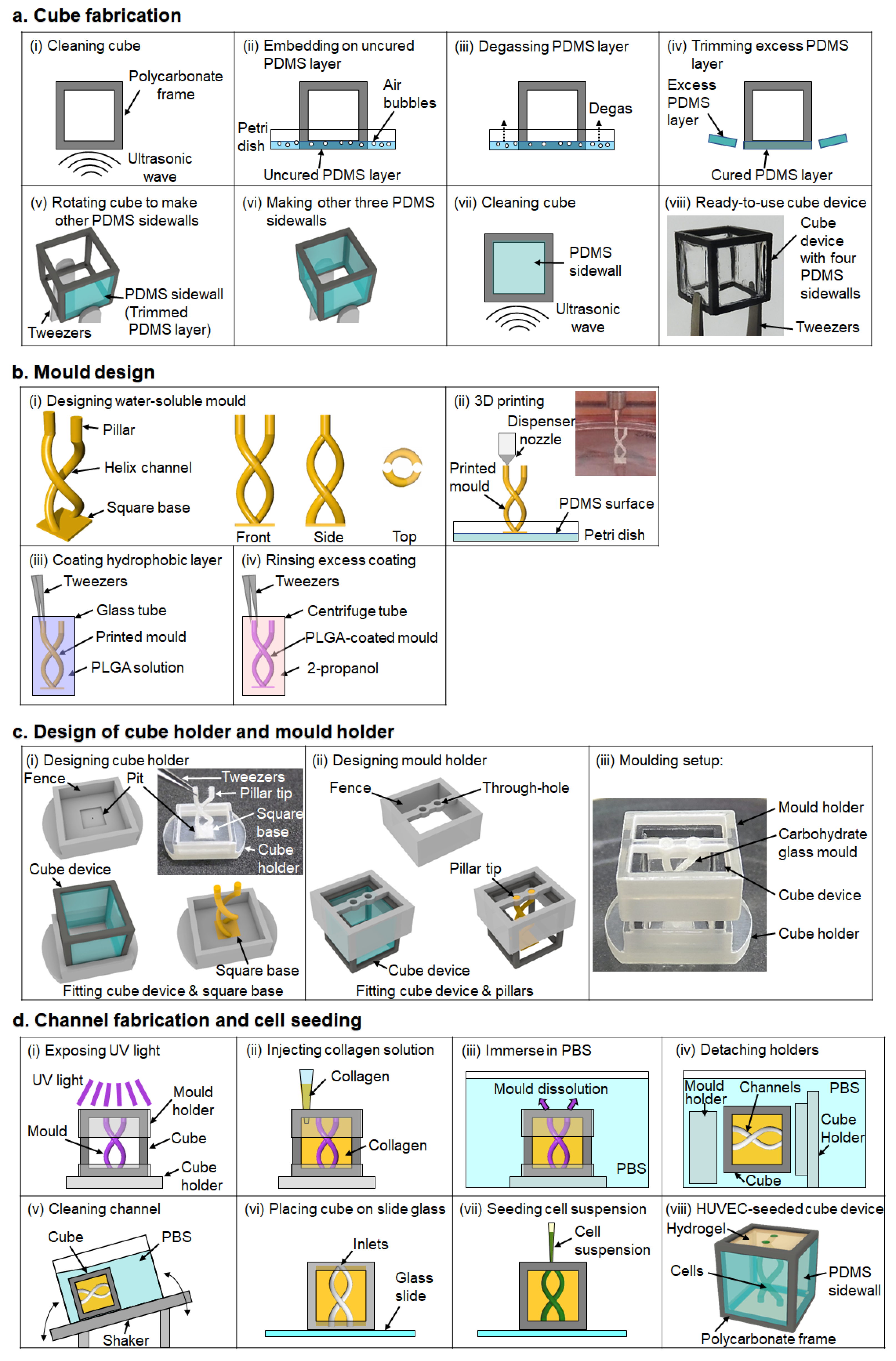
2.2. Preparation of Cube Device
A number of 10 mm-sized cube frames were made by machining. To make a PDMS sidewall, the frames were cleaned with 2-propanol followed by deionized water, for 10 min each with ultrasonication and then dried by blowing with compressed air. The frames were placed in an oven at 85 °C for 1 h to ensure the frames were free of moisture. The frames were placed on an uncured PDMS-coated surface of a 100 mm polystyrene dish. After the dish was degassed at −0.08 MPa for 30 min, the dish was placed in the oven at 85 °C for 1 h to cure the PDMS. The PDMS surface was trimmed with a scalpel along the frame to form the PDMS sidewall. The process was repeated three more times with the frame rotated 90° each time to cover the other three lateral surfaces with PDMS in the same manner.
2.3. 3D Printing of Water-Soluble Mold
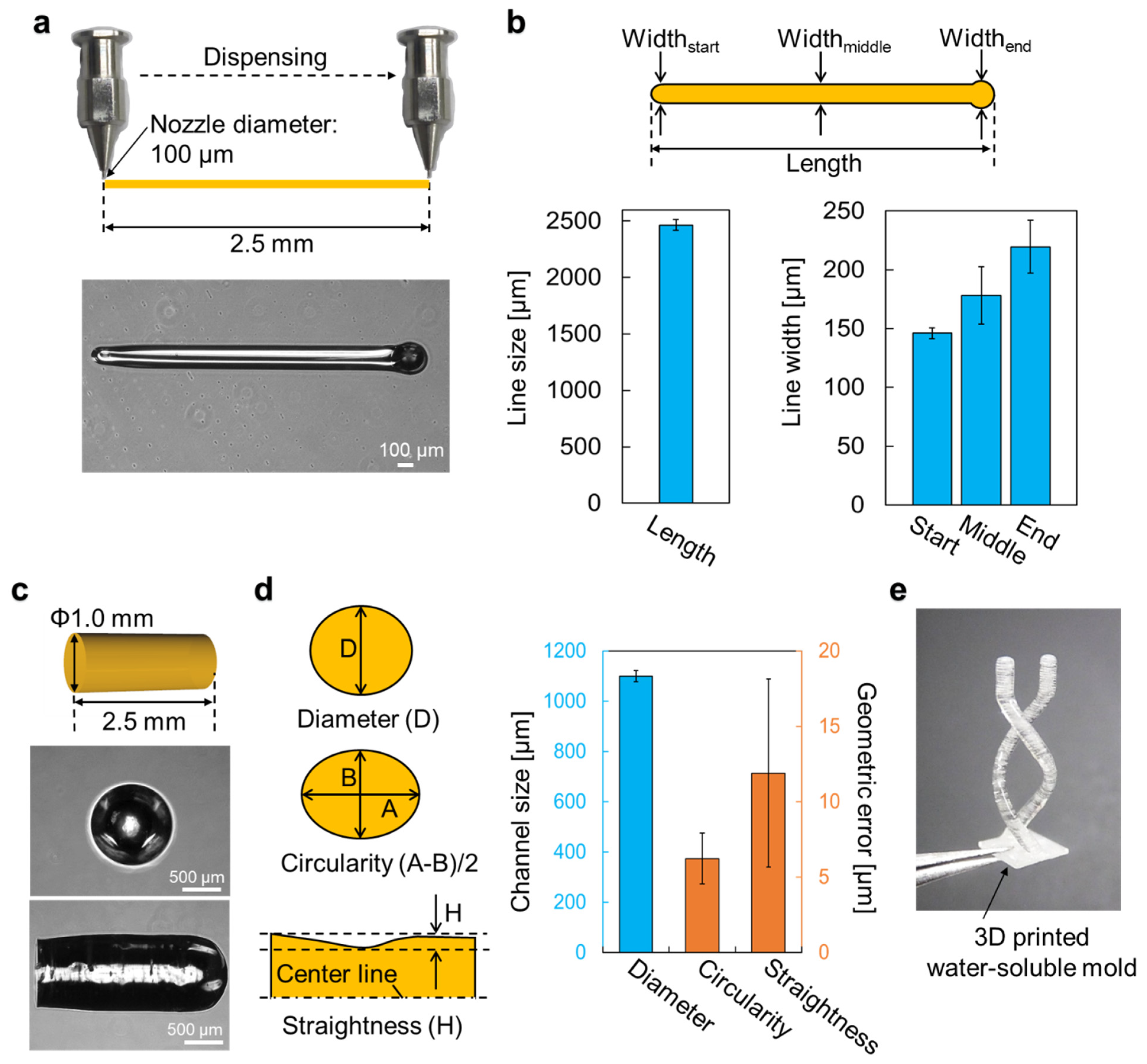
Molds were designed with a 3D modelling software (Rhinoceros 3D, McNeel, Seattle, WA, USA) and exported to G-codes with a 3D printing software (Repetier-host, Hot-World, Willich, Germany). The molds were then printed on a PDMS surface with a bioprinter (Allevi 1, Allevi, Philadelphia, PA, USA) using carbohydrate glass. Configurations of the bioprinter’s dispensing unit consisted of a 5 mL steel cylinder and a φ100 μm dispenser nozzle (SHN-0.1N, Musashi Engineering, Tokyo, Japan). To print the molds, optimized printing conditions of printing speed, discharge pressure, and printing temperature were 5 mm/s, 70–80 PSI, and 150 °C, respectively. Printed molds can be stored for a few days in a dehumidifying cabinet (ND-1S, AS One, Osaka, Japan) below 30% relative humidity to prevent the water-soluble molds from dissolving.
2.4. Hydrophobic Coating
Before hydrophobic coating, the molds were put in a cell culture CO2 incubator (5% pCO2, 95% humidity, 37 °C) for 30 min to smoothen the surface of the mold and then the molds were stored in the dry cabinet for a few hours to remove moisture from the mold. Once dry, the mold was immersed in 50 mg/mL PLGA solution for 3 min to make a hydrophobic coating on the mold. Immediately after that, the mold was immersed in 2-propanol for 1 min to remove excess PLGA, and 2-propanol was removed from the mold by blowing with compressed air. After the hydrophobic coating, the mold was bonded, using 5 µL of 50 mg/mL PLGA solution, to a cube holder that was printed using AR-M2 resin with a stereolithography 3D printer (Agilista 3200, Keyence, Osaka, Japan), then stored in the dry cabinet for at least 3 h to allow PLGA to adhere to the mold.
2.5. Molding
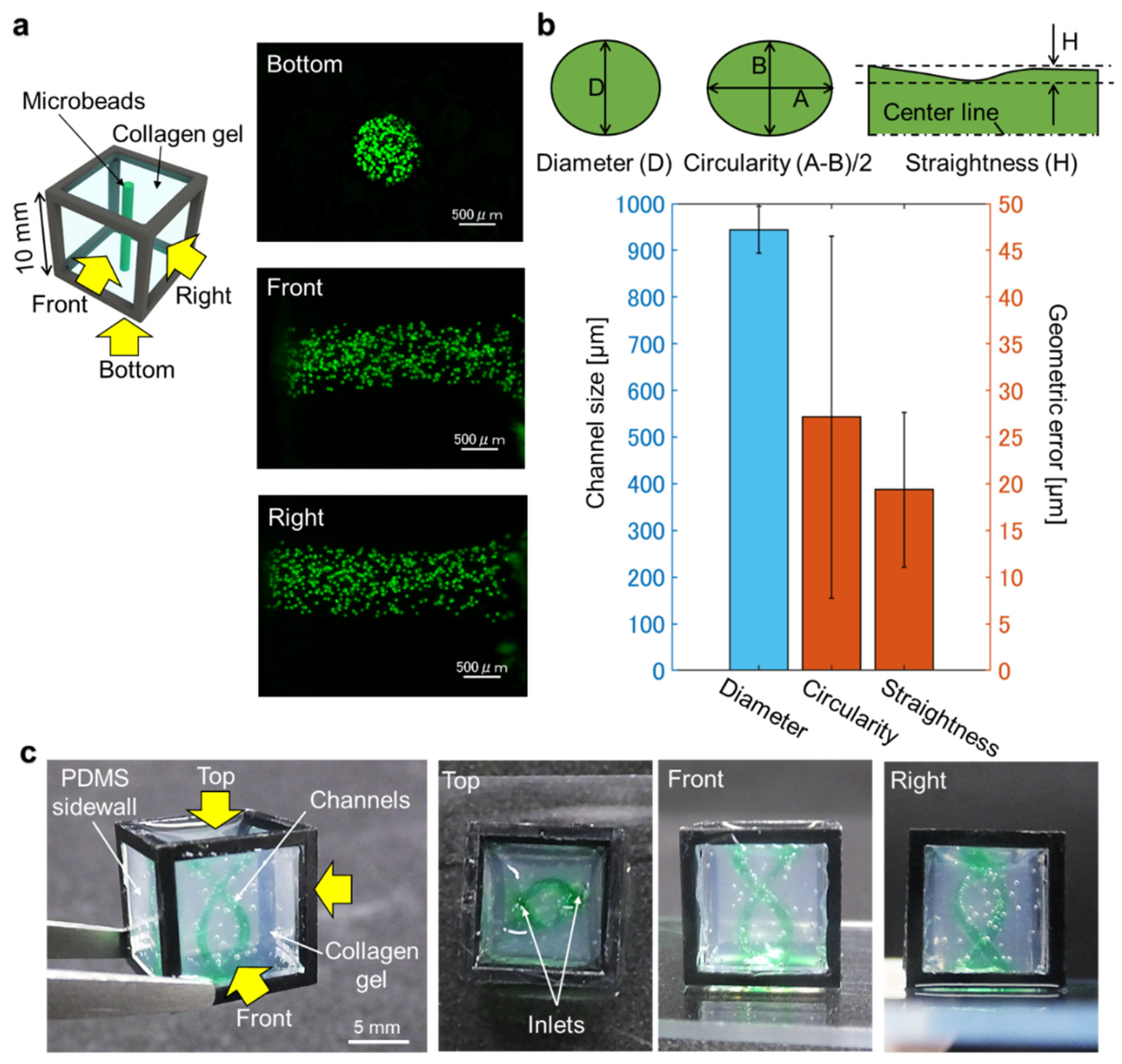
A mold holder was also 3D-printed using AR-M2 resin, to hold the mold in place in the cube. Before transferring the mold into the hydrogel, the cube device was exposed to vacuum plasma (−10 kPa, 3 min) with a plasma treater (PIB-10, vacuum device) to hydrophilize the cube device to prevent the generation of bubbles in the corners of the cube device when hydrogel is added into the cube. The molding setup in preparation for hydrogel injection was assembled together as three components: the hydrophobic coated mold attached to the cube holder on the bottom, the cube device placed on the cube holder, and the mold holder on top of the cube. Collagen solution prepared according to manufacturer’s protocol at 4 °C was then injected into the cube device from the top side of the setup. After curing the collagen for 30 min on a hotplate at 37 °C, the setup was immersed in PBS to dissolve the water-soluble mold for at least 90 min in a CO2 incubator. Then, the cube holder and mold holder were removed from the cube device while still immersed in PBS bath, before being transferred to fresh PBS. The cube in PBS was placed on a see-saw shaker (NA-M101, Nissin, Tokyo, Japan) for 10 min to wash out residual mold.
2.6. Cell Seeding
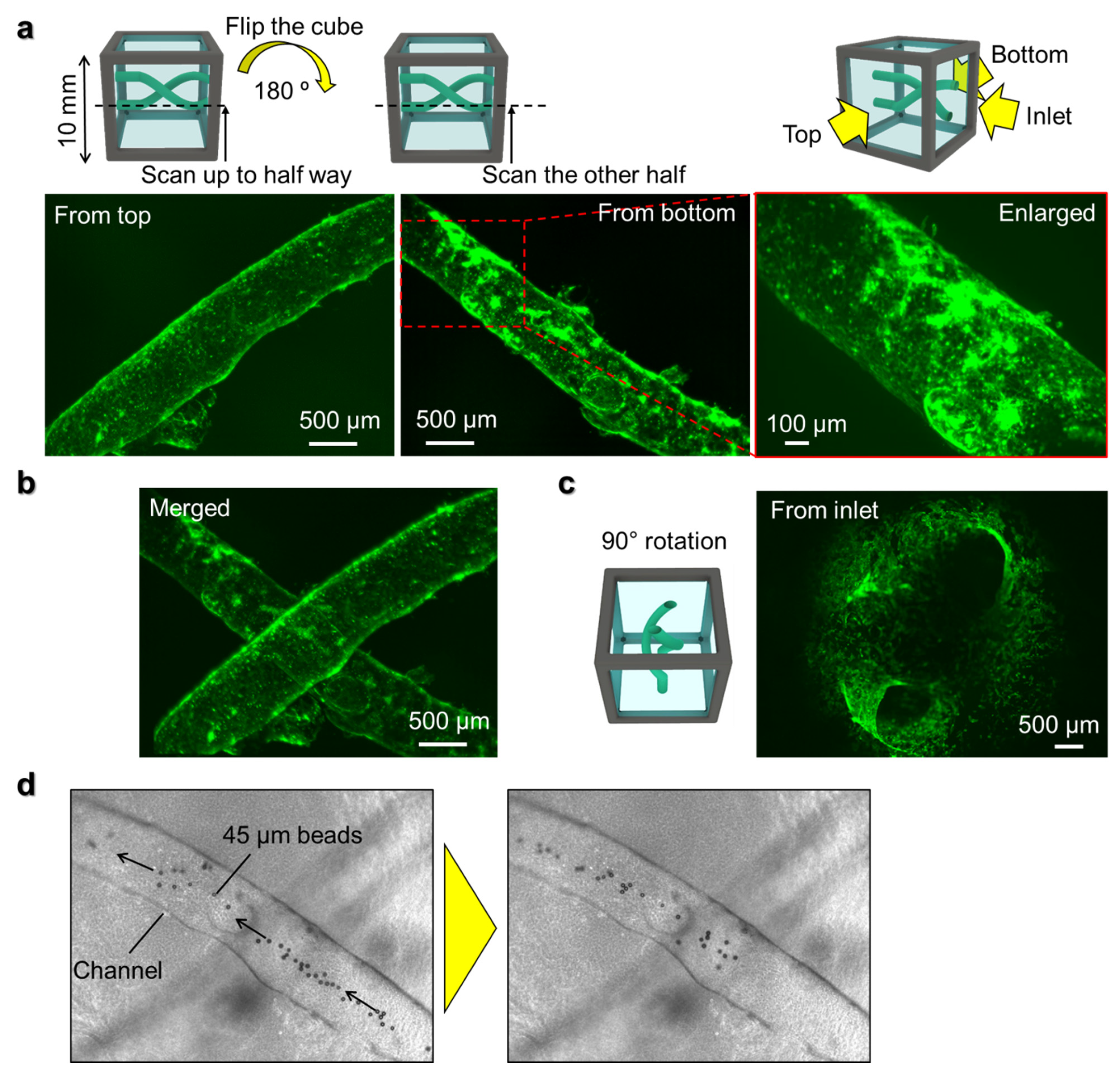
HUVEC was cultured in EGM-2 and used at passage 4. To prepare the cell suspension of HUVEC, HUVEC around 80% confluency was washed with PBS, then treated with Trypsin-EDTA for 2.5 min at 37 °C before neutralizing the trypsin with TNS. Collected HUVEC was centrifuged at 300× g, 4 °C for 4 min. After aspirating the supernatant, cell density was adjusted to 30 × 106 cells/mL using EGM-2. Before cell seeding, the cube device with the inlets of channels facing up was placed on a glass slide and the outer surfaces of the cube device were wiped gently with a Kimwipe wipe to remove excess moisture. An amount of 10 µL of the HUVEC suspension was injected into the inlets of the channels. After confirming that the HUVEC suspension had flowed through the channel, the cube device was rotated 90° and transferred to a 12-well plate. The cube device was incubated in a CO2 incubator for 30 min to allow the HUVEC to adhere to the inner wall of the channels. Then, the cube device was rotated 180° and incubated for another 30 min for the HUVEC to adhere to the opposite wall of the channel. The rotation and incubation were repeated twice in the same way. Then, the cube device, with the inlets facing up, was placed on a slide glass again and another 10 µL of fresh HUVEC suspension at 30 × 106 cells/mL was injected and the rotation-incubation procedure repeated to enable the HUVEC to adhere to the two remaining surfaces of the channel, so that the entire inner wall of the channel is seeded with cells. After seeding, the cells were cultured for 5 days in 2 mL of EGM-2 in a 24-well plate on a see-saw shaker in a CO2 incubator. Every day, the cube device was rotated 90°, and 1 mL of EGM-2 was replaced with fresh medium.
2.7. Immunofluorescence Staining
Samples were washed with PBS for 5 min and fixed with 4% PFA for 20 min at room temperature. Then, the samples were washed with PBS for 5 min at room temperature before permeabilization with 0.5% Triton X-100 for 10 min at 4 °C. After rinsing with PBS three times, the samples were treated with 100 mM glycine for 15 min at room temperature and rinsed once with PBS. Blocking was performed for 45 min at room temperature with IF-buffer (10% Goat serum, 0.2% Triton X-100, 0.1% BSA, and 0.05% Tween-20 in PBS), and then stained with Alexa fluor 488 Phalloidin (1:200) for 45 min at room temperature. The samples were then washed three times with PBS for 5 min at room temperature before imaging.
2.8. Measurement of Water-Soluble Molds
3D-printed molds were imaged with a phase-contrast microscope (CKX41, Olympus, Waltham, MA, USA). Dimensions of the molds were measured from the acquired phase-contrast images with open-source image processing software (ImageJ, NIH, Bethesda, MD, USA).
2.9. Measurement of Channels in Gel
Polystyrene-latex fluorescent beads (45 µm diameter, excitation wavelength = 441.53 nm, emission wavelength = 485.56 nm) diluted in 1.5% agarose solution was injected into the channels formed in the collagen gel in the cube device and cooled for 3 min at 4 °C. Z-stack images of the channels were taken from multiple directions with a fluorescence microscope (BZX-700, Keyence, Osaka, Japan) and z-projection images were exported to ImageJ for measurement analyses.
2.10. Multi-Directional Imaging
Z-stack images of fluorescently stained HUVEC in the cube device were taken from multiple directions with the fluorescence microscope with sectioning function by rotating the cube device and taking images from each of its sides. The z-stack images were overlaid with an image alignment software that was developed in-house and then exported as z-projection images using ImageJ.
4. Discussion and Conclusions
The method detailed in this paper enables the control of cell position with an unrestricted range of 3D shapes in hydrogel in a cube device by using a water-soluble mold, which we demonstrated by forming a helix-shaped vascular structure using human endothelial cells. By scanning the structure from multiple angles, large-scale high-resolution images of the whole sample could be obtained using a simple fluorescence microscope with a low magnification lens. Thus, the platform developed here has high potential to contribute to the field of organoids by offering control on organoid morphogenesis and high-resolution imaging of millimeter-sized organoids.
Current organoid culture methods mainly rely on culturing cells as a simple spheroid or cluster of cells and allowing the cells to self-organize into organoids based on specific biochemical cues from the differentiating factors in the medium. Nevertheless, the shape and geometry of the surrounding microenvironment also affect cellular migration, proliferation, and differentiation, as cells collectively react to the differential morphogen signaling gradients and mechanical stresses caused by the geometrical constraints exerted on them [
19,
20]. Hence, the ability to control cell shape according to the shape of the target organ may contribute to the development of more sophisticated organoids such as lung organoids with highly controlled branching formations or kidney organoids with nephrons precisely formed with the Bowman’s capsule on one end and the collecting duct on the other.
One of the remaining challenges with this method, however, is that some of the hydrophobic coating of the mold remains on the inner walls of the formed shape, and some cells may preferentially adhere to the PLGA surface rather than the hydrogel. Although PLGA is a biocompatible material and has been used in various applications such as in bone and kidney regeneration or as drug delivery carriers [
21,
22,
23], it is still unknown if the residual PLGA may have an effect on the differentiation of stem cells such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) that are used in organoid generation, particularly as most organoid culture protocols are based on the use of soft hydrogels such as Matrigel. Reducing the concentration and coating time of the PLGA coating solution makes it easier to eliminate the PLGA residues, but doing so will compromise the accuracy and reproducibility of the shape formed in the hydrogel. Therefore, it is necessary to consider how to remove the hydrophobic coating after transferring the mold shape to the hydrogel without using harsh treatments that may cause damage to the gel or the cells. Some potential methods for removing residual PLGA are by cleaning with hydrolytic enzymes that degrade PLGA [
24] after the mold has been dissolved, or by pre-mixing the degrading enzyme with the coating solution as was reported for poly(ε-caprolactone) (PCL), which is another popular hydrophobic coating polymer [
25]. The optimization of such residue removal protocols for the platform developed here is currently ongoing. Another major challenge is the resolution of the mold, which depends on the diameter of the dispensing nozzle. In this research, a nozzle with a diameter of 100 µm was used, but molds with higher resolution could be attained if a nozzle with a smaller diameter was used. However, if the nozzle diameter is too small, it becomes difficult to dispense highly viscous inks such as carbohydrate glass, so the trade-off between reducing the diameter of the nozzle for high-resolution printing and printability needs to be considered.
The concept of the 3D culture platform developed in this study shows the feasibility of designing organoids with complex shapes, and the potential to analyze the morphogenetic mechanisms of organoid formation by controlling the initial position of cells. It is expected that this technology will provide new ways to develop organoids with more complex structures and functions, which can contribute to a wide range of fields such as developmental biology, drug development, and regenerative medicine.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




