



Antioxidant Dimethyl Fumarate Temporarily but Not Chronically Improves Intracortical Microelectrode Performance

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
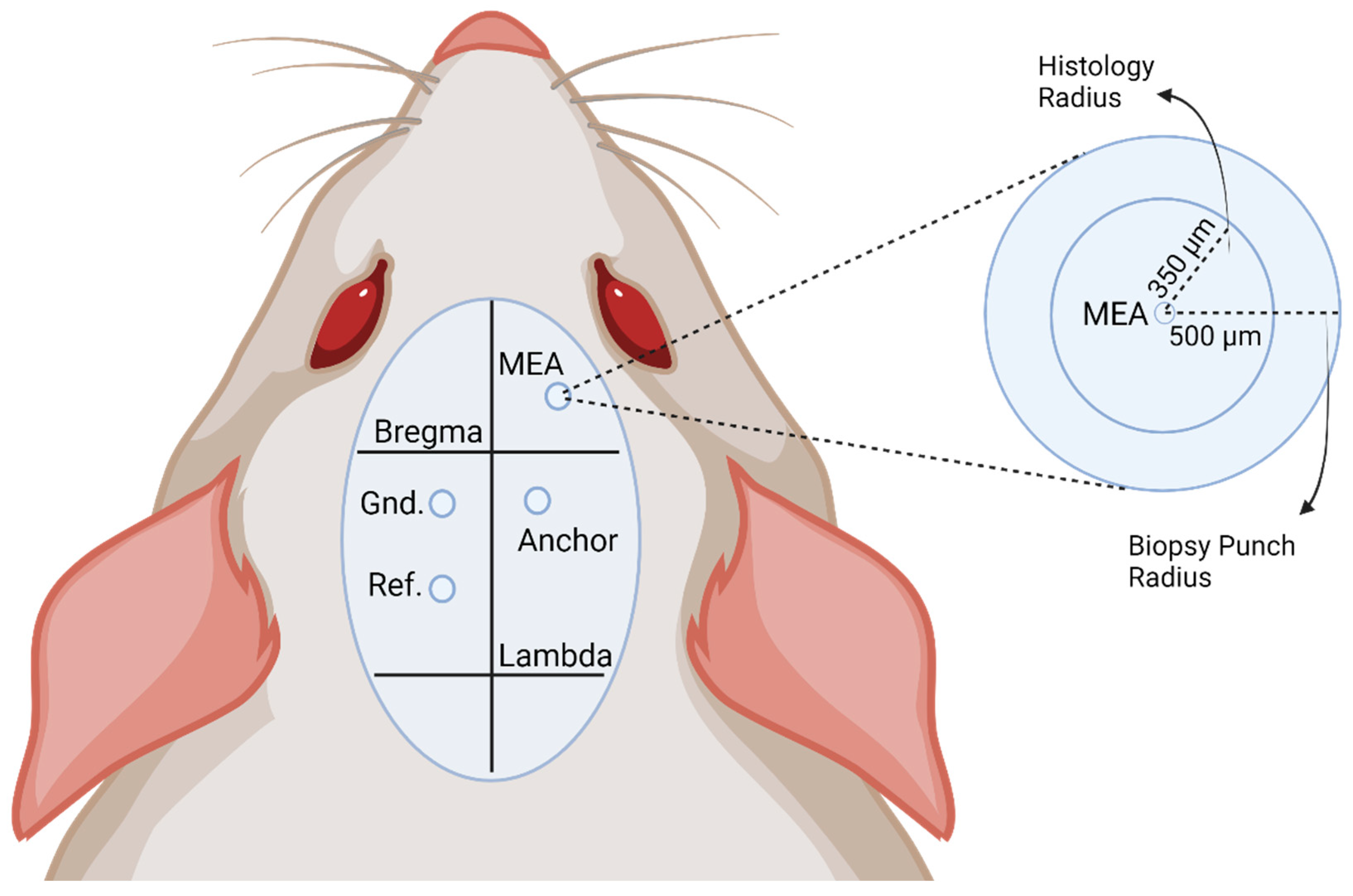
2.1. Intracortical Microelectrode Array Preparation
2.2. Animals and Surgical Implantation
2.3. Drug Preparation and Delivery
2.4. Neurophysiological Recordings
2.5. Neurophysiological Analysis
2.6. Histological Tissue Processing
2.7. Immunohistochemical Staining
2.8. Imaging and Analysis
2.9. Bulk Gene Analysis
2.10. Statistical Analysis
3. Results
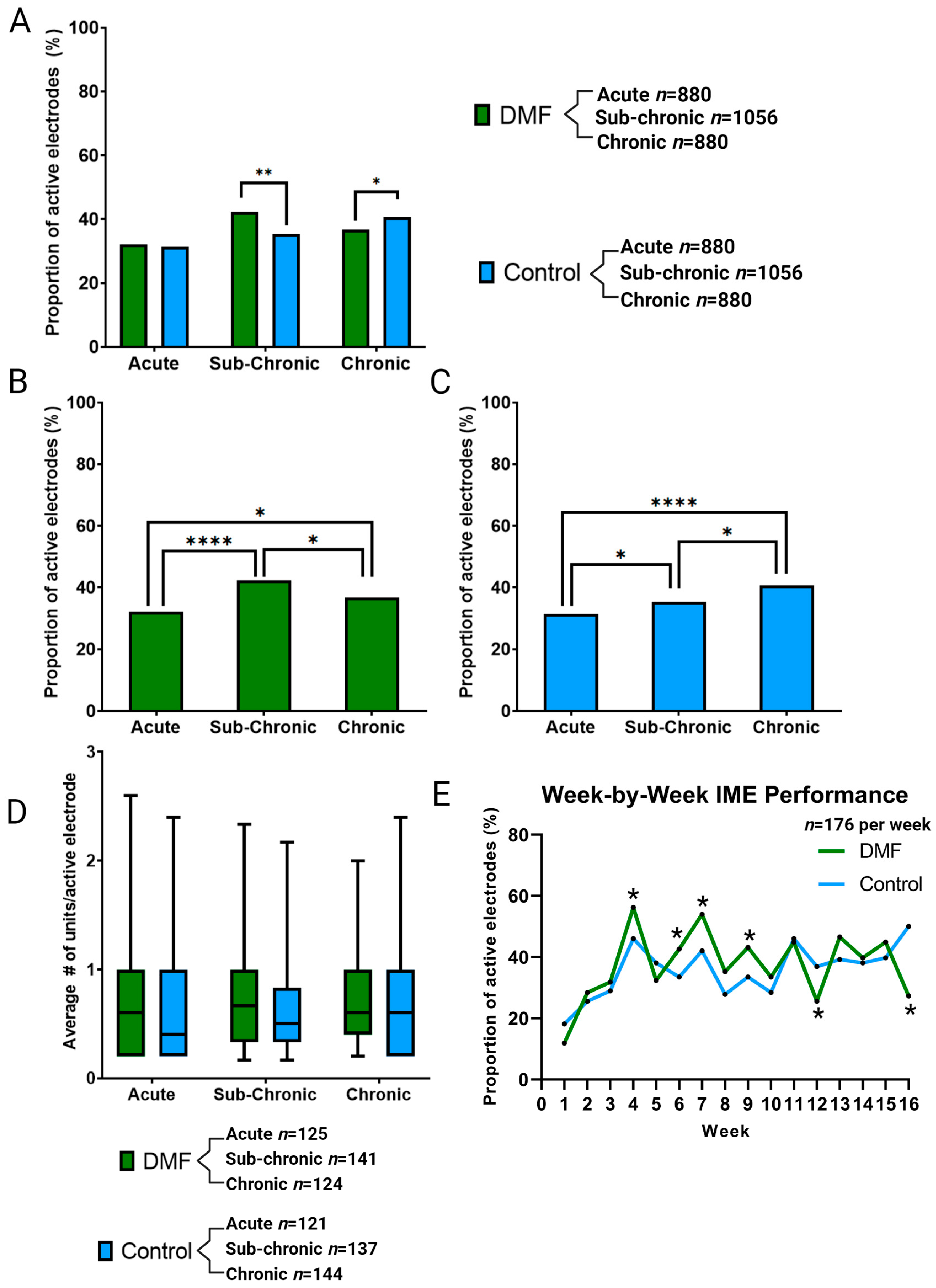
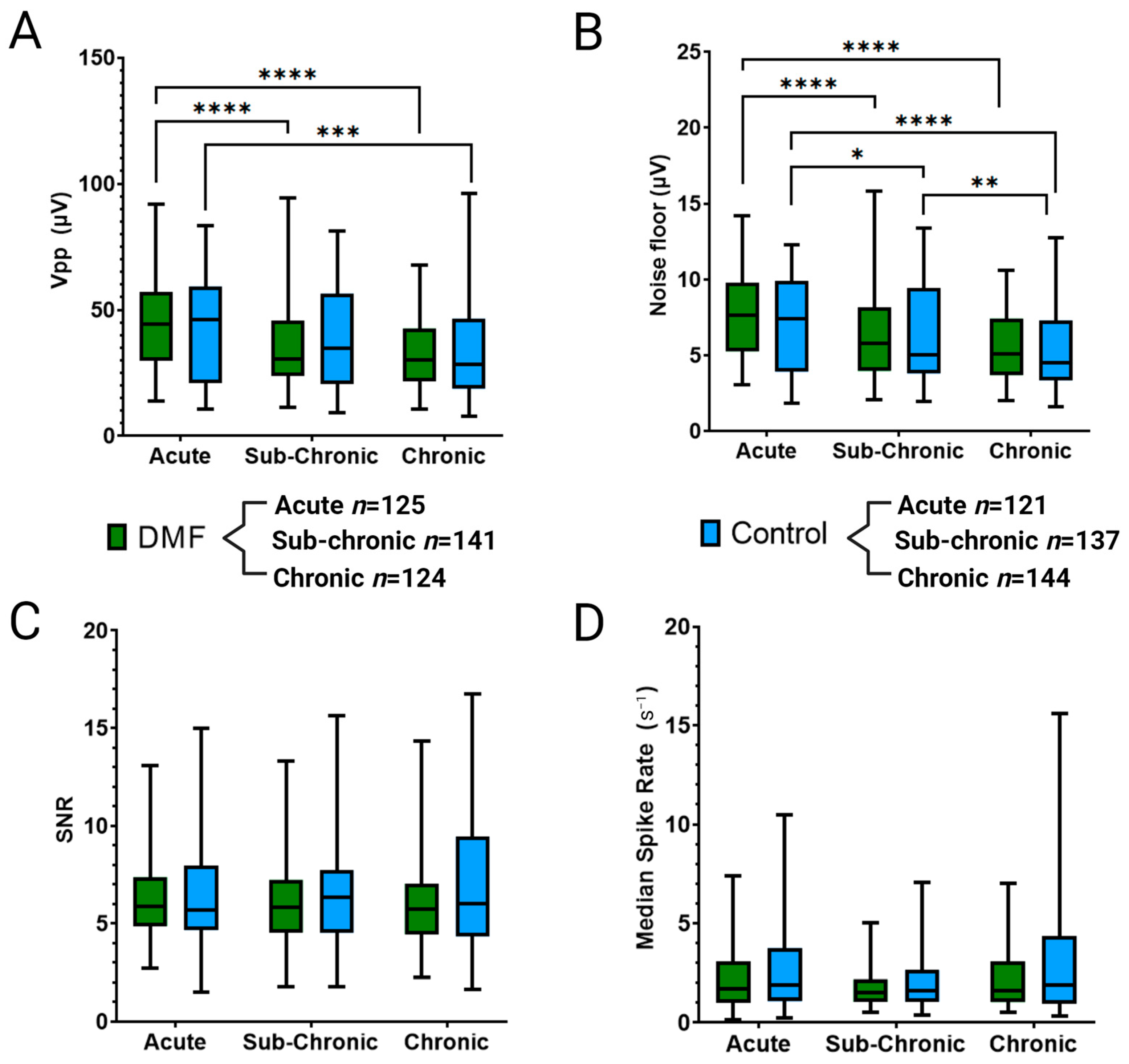
3.1. Effect of DMF on MEA Performance and Single-Unit Recordings
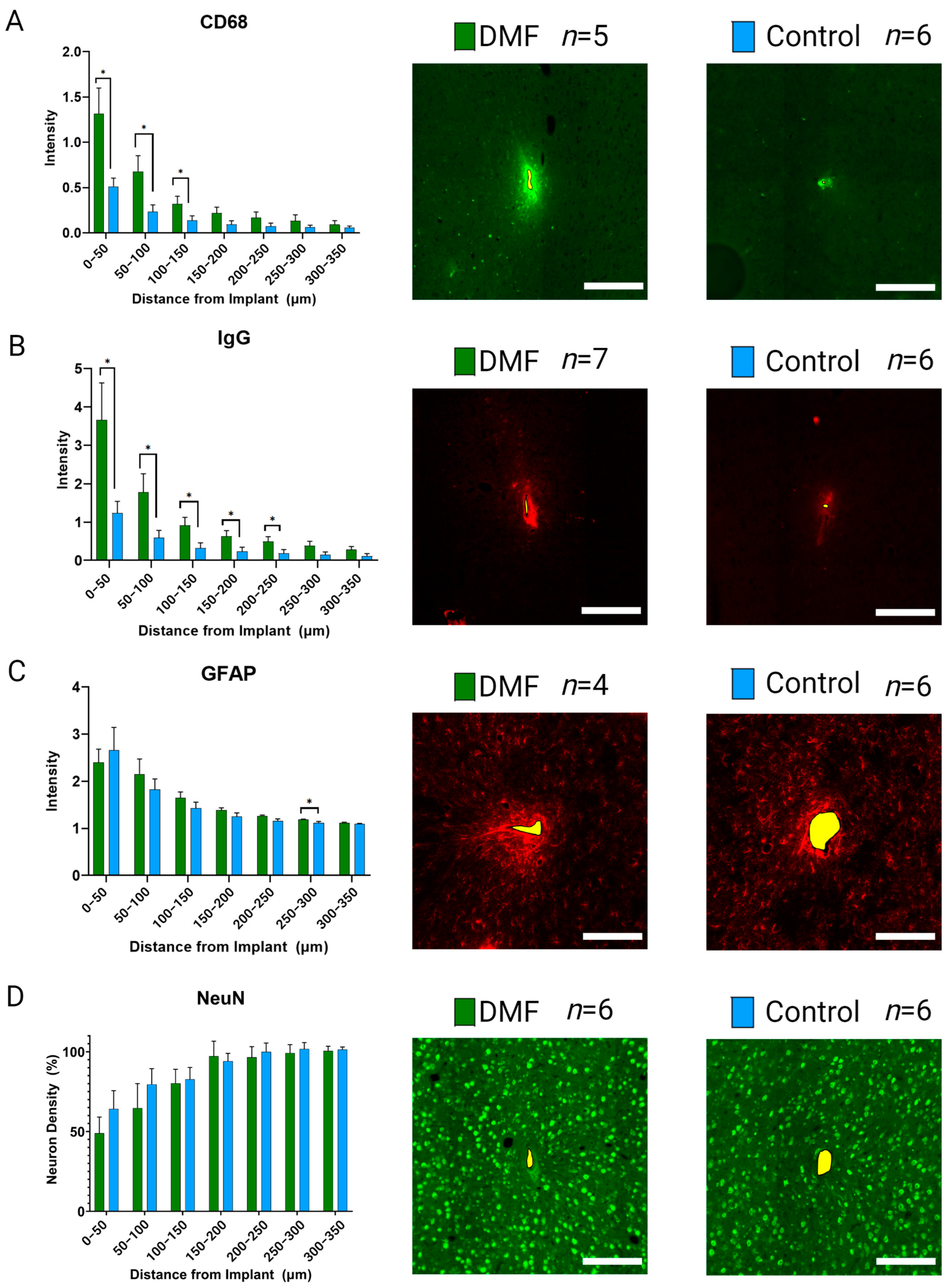
3.2. Effect of DMF on Tissue Health and Neuroinflammation
3.2.1. Chronic Inflammation and Blood-Brain Barrier Permeability
3.2.2. Neural Health and Viability
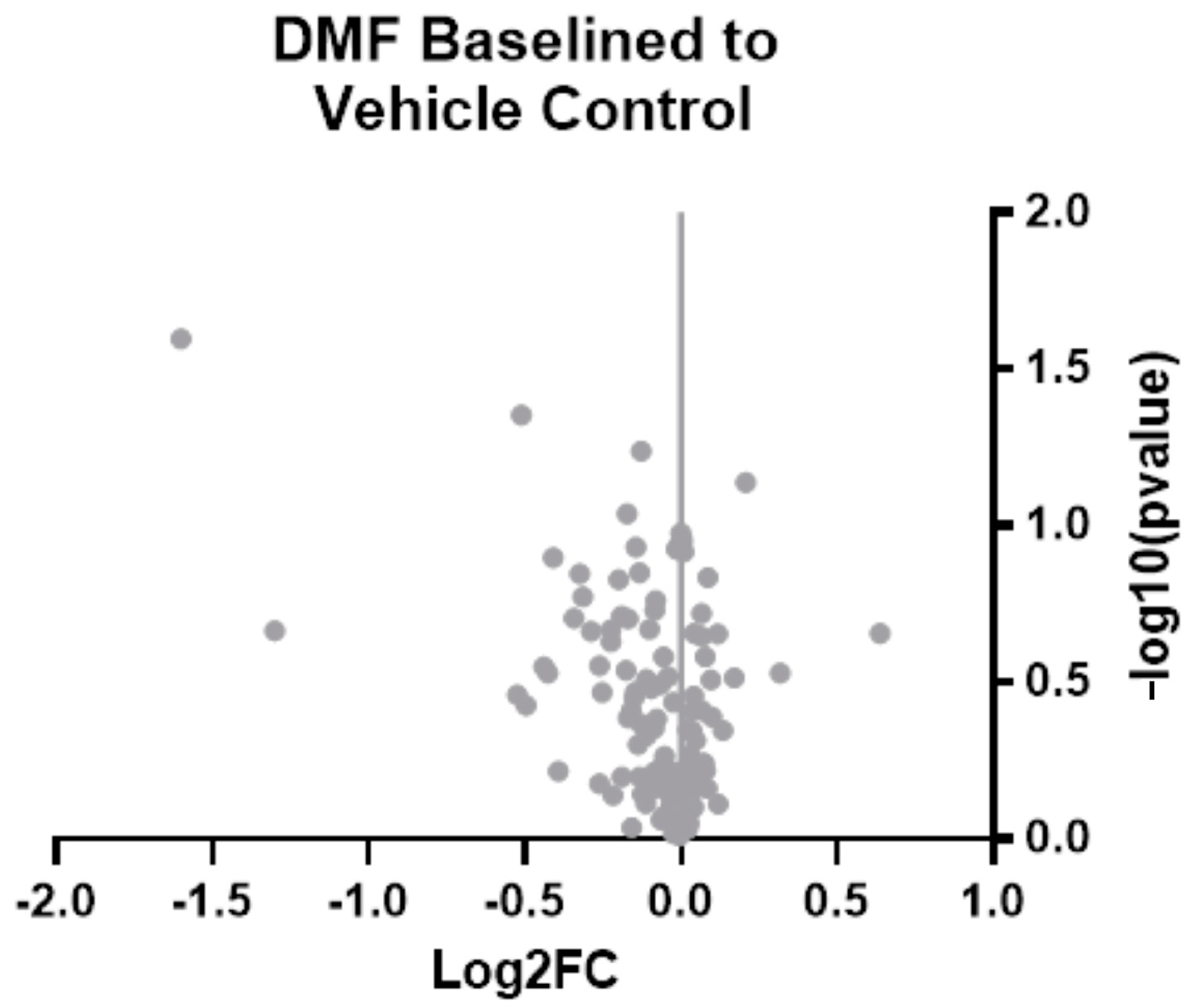
3.3. Bulk Gene Expression Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Simeral, J.D.; Kim, S.P.; Black, M.J.; Donoghue, J.P.; Hochberg, L.R. Neural control of cursor trajectory and click by a human with tetraplegia 1000 days after implant of an intracortical microelectrode array. J. Neural Eng. 2011, 8, 025027. [Google Scholar] [CrossRef] [PubMed]
- Vilela, M.; Hochberg, L.R. Applications of brain-computer interfaces to the control of robotic and prosthetic arms. Handb. Clin. Neurol. 2020, 168, 87–99. [Google Scholar] [CrossRef]
- Shih, J.J.; Krusienski, D.J.; Wolpaw, J.R. Brain-computer interfaces in medicine. Mayo Clin. Proc. 2012, 87, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Fatima, N.; Shuaib, A.; Saqqur, M. Intra-cortical brain-machine interfaces for controlling upper-limb powered muscle and robotic systems in spinal cord injury. Clin. Neurol. Neurosurg. 2020, 196, 106069. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Eguren, D.; Azorin, J.M.; Grossman, R.G.; Luu, T.P.; Contreras-Vidal, J.L. Brain-machine interfaces for controlling lower-limb powered robotic systems. J. Neural Eng. 2018, 15, 021004. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, M.; Skousen, J.L.; Weder, C.; Capadona, J.R. Progress Towards Biocompatible Intracortical Microelectrodes for Neural Interfacing Applications. J. Neural Eng. 2015, 12, 011001. [Google Scholar] [CrossRef]
- Kozai, T.D.; Jaquins-Gerstl, A.S.; Vazquez, A.L.; Michael, A.C.; Cui, X.T. Brain tissue responses to neural implants impact signal sensitivity and intervention strategies. ACS Chem. Neurosci. 2015, 6, 48–67. [Google Scholar] [CrossRef]
- Wellman, S.M.; Li, L.; Yaxiaer, Y.; McNamara, I.; Kozai, T.D.Y. Revealing Spatial and Temporal Patterns of Cell Death, Glial Proliferation, and Blood-Brain Barrier Dysfunction Around Implanted Intracortical Neural Interfaces. Front. Neurosci. 2019, 13, 493. [Google Scholar] [CrossRef]
- Barrese, J.C.; Rao, N.; Paroo, K.; Triebwasser, C.; Vargas-Irwin, C.; Franquemont, L.; Donoghue, J.P. Failure mode analysis of silicon-based intracortical microelectrode arrays in non-human primates. J. Neural Eng. 2013, 10, 066014. [Google Scholar] [CrossRef]
- Freire, M.A.; Morya, E.; Faber, J.; Santos, J.R.; Guimaraes, J.S.; Lemos, N.A.; Sameshima, K.; Pereira, A.; Ribeiro, S.; Nicolelis, M.A. Comprehensive analysis of tissue preservation and recording quality from chronic multielectrode implants. PLoS ONE 2011, 6, e27554. [Google Scholar] [CrossRef]
- Bennett, C.; Mohammed, F.; Alvarez-Ciara, A.; Nguyen, M.A.; Dietrich, W.D.; Rajguru, S.M.; Streit, W.J.; Prasad, A. Neuroinflammation, oxidative stress, and blood-brain barrier (BBB) disruption in acute Utah electrode array implants and the effect of deferoxamine as an iron chelator on acute foreign body response. Biomaterials 2019, 188, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.; Samikkannu, M.; Mohammed, F.; Dietrich, W.D.; Rajguru, S.M.; Prasad, A. Blood brain barrier (BBB)-disruption in intracortical silicon microelectrode implants. Biomaterials 2018, 164, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.A.; Buck, A.C.; Self, W.K.; Capadona, J.R. Stab injury and device implantation within the brain results in inversely multiphasic neuroinflammatory and neurodegenerative responses. J. Neural Eng. 2012, 9, 046020. [Google Scholar] [CrossRef] [PubMed]
- Polikov, V.; Tresco, P.; Reichert, W. Response of brain tissue to chronically implanted neural electrodes. J. Neurosci. Methods 2005, 148, 1–18. [Google Scholar] [CrossRef]
- Chen, K.; Wellman, S.M.; Yaxiaer, Y.; Eles, J.R.; Kozai, T.D.Y. In vivo spatiotemporal patterns of oligodendrocyte and myelin damage at the neural electrode interface. Biomaterials 2021, 268, 120526. [Google Scholar] [CrossRef]
- Kozai, T.D.Y.; Marzullo, T.C.; Hooi, F.; Langhals, N.B.; Majewska, A.K.; Brown, E.B.; Kipke, D.R. Reduction of neurovascular damage resulting from microelectrode insertion into the cerebral cortex using in vivo two-photon mapping. J. Neural Eng. 2010, 7, 046011. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, M.; Sunil, S.; Black, J.; Barkauskas, D.; Haung, A.Y.; Miller, R.H.; Selkirk, S.M.; Capadona, J.R. The Roles of Blood-derived Macrophages and Resident Microglia in the Neuroinflammatory Response to Implanted Intracortical Microelectrodes. Biomaterials 2014, 35, 8049–8064. [Google Scholar] [CrossRef]
- Bedell, H.W.; Hermann, J.K.; Ravikumar, M.; Lin, S.; Rein, A.; Li, X.; Molinich, E.; Smith, P.D.; Selkirk, S.M.; Miller, R.H. Targeting CD14 on blood derived cells improves intracortical microelectrode performance. Biomaterials 2018, 163, 163–173. [Google Scholar] [CrossRef]
- Bedell, H.W.; Song, S.; Li, X.; Molinich, E.; Lin, S.; Voit, W.E.; Pancrazio, J.J.; Capadona, J.R. Understanding the effects of both CD14-meditated innate immunity and device/tissue mechanical mismatch in the neuroinflammatory response to intracortical microelectrodes. Front. Neurosci. 2018, 12, 772. [Google Scholar] [CrossRef]
- Bedell, H.W.; Capadona, J.R. Anti-inflammatory Approaches to Mitigate the Neuroinflammatory Response to Brain-Dwelling Intracortical Microelectrodes. J. Immunol. Sci. 2018, 2, 15–21. [Google Scholar] [CrossRef]
- Hermann, J.K.; Ravikumar, M.; Shoffstall, A.J.; Ereifej, E.S.; Kovach, K.M.; Chang, J.; Soffer, A.; Wong, C.; Srivastava, V.; Smith, P.; et al. Inhibition of the cluster of differentiation 14 innate immunity pathway with IAXO-101 improves chronic microelectrode performance. J. Neural Eng. 2018, 15, 025002. [Google Scholar] [CrossRef] [PubMed]
- Hermann, J.K.; Lin, S.; Soffer, A.; Wong, C.; Srivastava, V.; Chang, J.; Sunil, S.; Sudhakar, S.; Tomaszewski, W.H.; Protasiewicz, G.; et al. The Role of Toll-Like Receptor 2 and 4 Innate Immunity Pathways in Intracortical Microelectrode-Induced Neuroinflammation. Front. Bioeng. Biotechnol. 2018, 6, 113. [Google Scholar] [CrossRef]
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Semin. Immunol. 2008, 20, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Potter-Baker, K.A.; Stewart, W.G.; Tomaszewski, W.H.; Wong, C.T.; Meador, W.D.; Ziats, N.P.; Capadona, J.R. Implications of chronic daily anti-oxidant administration on the inflammatory response to intracortical microelectrodes. J. Neural Eng. 2015, 12, 046002. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.A.; Jorfi, M.; Householder, K.T.; Foster, E.J.; Weder, C.; Capadona, J.R. Curcumin-releasing mechanically-adaptive intracortical implants improve the proximal neuronal density and blood-brain barrier stability. Acta Biomater. 2014, 10, 2209–2222. [Google Scholar] [CrossRef]
- Potter, K.A.; Buck, A.C.; Self, W.K.; Callanan, M.E.; Sunil, S.; Capadona, J.R. The effect of resveratrol on neurodegeneration and blood brain barrier stability surrounding intracortical microelectrodes. Biomaterials 2013, 34, 7001–7015. [Google Scholar] [CrossRef]
- Ereifej, E.S.; Rial, G.M.; Hermann, J.K.; Smith, C.S.; Meade, S.M.; Rayyan, J.M.; Chen, K.; Feng, H.; Capadona, J.R. Implantation of Neural Probes in the Brain Elicits Oxidative Stress. Front. Bioeng. Biotechnol. 2018, 6, 9. [Google Scholar] [CrossRef]
- Potter-Baker, K.A.; Capadona, J.R. Reducing the “Stress”: Antioxidative Therapeutic and Material Approaches May Prevent Intracortical Microelectrode Failure. ACS Macro Lett. 2015, 4, 275–279. [Google Scholar] [CrossRef]
- Salatino, J.W.; Ludwig, K.A.; Kozai, T.D.Y.; Purcell, E.K. Glial responses to implanted electrodes in the brain. Nat. Biomed. Eng. 2017, 1, 862–877. [Google Scholar] [CrossRef]
- Spencer, K.C.; Sy, J.C.; Falcon-Banchs, R.; Cima, M.J. A three dimensional in vitro glial scar model to investigate the local strain effects from micromotion around neural implants. Lab Chip 2017, 17, 795–804. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell. Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef]
- Colton, C.A.; Chernyshev, O.N.; Gilbert, D.L.; Vitek, M.P. Microglial contribution to oxidative stress in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 899, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Ding, S.J.; Wu, T.; Zhu, Y.L. Correlation of free radical level and apoptosis after intracerebral hemorrhage in rats. Neurosci. Bull. 2008, 24, 351–358. [Google Scholar] [CrossRef]
- Katsu, M.; Niizuma, K.; Yoshioka, H.; Okami, N.; Sakata, H.; Chan, P.H. Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. J. Cereb. Blood Flow Metab. 2010, 30, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ereifej, E.S.; Schwartzman, W.E.; Meade, S.M.; Chen, K.; Rayyan, J.; Feng, H.; Aluri, V.; Mueller, N.N.; Bhambra, R.; et al. Investigation of the Feasibility of Ventricular Delivery of Resveratrol to the Microelectrode Tissue Interface. Micromachines 2021, 12, 1446. [Google Scholar] [CrossRef]
- Potter-Baker, K.A.; Nguyen, J.K.; Kovach, K.M.; Gitomer, M.M.; Srail, T.W.; Stewart, W.G.; Skousen, J.L.; Capadona, J.R. Development of Superoxide Dismutase Mimetic Surfaces to Reduce Accumulation of Reactive Oxygen Species Surrounding Intracortical Microelectrodes. J. Mater. Chem. B 2014, 2, 2248–2258. [Google Scholar] [CrossRef]
- Hernandez-Reynoso, A.G.; Sturgill, B.; Hoeferlin, G.F.; Druschel, L.D.; Krebs, O.K.; Menendez, D.; Thai, T.T.D.; Smith, T.J.; Duncan, J.; Zhang, J.; et al. The Effect of a Mn(III)tetrakis(4- benzoic acid)porphyrin (MnTBAP) Coating on the Chronic Recording Performance of Planar Silicon Intracortical Microelectrode Arrays. Biomaterials, 2023; under review. [Google Scholar]
- Biogen Idec. Tecfidera (Dimethyl Fumarate): US Prescribing Information; Biogen Idec: Cambridge, MA, USA, 2013. [Google Scholar]
- United States Food and Drug Administration. TECFIDERA™ Prescribing Information; United States Food and Drug Administration: Silver Spring, MD, USA, 2013. [Google Scholar]
- Casili, G.; Campolo, M.; Paterniti, I.; Lanza, M.; Filippone, A.; Cuzzocrea, S.; Esposito, E. Dimethyl Fumarate Attenuates Neuroinflammation and Neurobehavioral Deficits Induced by Experimental Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1437–1451. [Google Scholar] [CrossRef]
- Campolo, M.; Casili, G.; Biundo, F.; Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Esposito, E. The Neuroprotective Effect of Dimethyl Fumarate in an MPTP-Mouse Model of Parkinson’s Disease: Involvement of Reactive Oxygen Species/Nuclear Factor-kappaB/Nuclear Transcription Factor Related to NF-E2. Antioxid. Redox Signal. 2017, 27, 453–471. [Google Scholar] [CrossRef]
- Cerina, M.; Narayanan, V.; Delank, A.; Meuth, P.; Graebenitz, S.; Gobel, K.; Herrmann, A.M.; Albrecht, S.; Daldrup, T.; Seidenbecher, T.; et al. Protective potential of dimethyl fumarate in a mouse model of thalamocortical demyelination. Brain Struct. Funct. 2018, 223, 3091–3106. [Google Scholar] [CrossRef]
- Gafson, A.R.; Savva, C.; Thorne, T.; David, M.; Gomez-Romero, M.; Lewis, M.R.; Nicholas, R.; Heslegrave, A.; Zetterberg, H.; Matthews, P.M. Breaking the cycle: Reversal of flux in the tricarboxylic acid cycle by dimethyl fumarate. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e562. [Google Scholar] [CrossRef]
- Kunze, R.; Urrutia, A.; Hoffmann, A.; Liu, H.; Helluy, X.; Pham, M.; Reischl, S.; Korff, T.; Marti, H.H. Dimethyl fumarate attenuates cerebral edema formation by protecting the blood-brain barrier integrity. Exp. Neurol. 2015, 266, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, S.; Tortorella, C.; Gasperini, C. Pharmacology and clinical efficacy of dimethyl fumarate (BG-12) for treatment of relapsing-remitting multiple sclerosis. Ther. Clin. Risk Manag. 2014, 10, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Saha, L.; Kumari, P.; Singh, J.; Bhatia, A.; Banerjee, D.; Chakrabarti, A. Effect of dimethyl fumarate on neuroinflammation and apoptosis in pentylenetetrazol kindling model in rats. Brain Res. Bull. 2019, 144, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Abbas, K.; Breton, J.; Planson, A.G.; Bouton, C.; Bignon, J.; Seguin, C.; Riquier, S.; Toledano, M.B.; Drapier, J.C. Nitric oxide activates an Nrf2/sulfiredoxin antioxidant pathway in macrophages. Free Radic. Biol. Med. 2011, 51, 107–114. [Google Scholar] [CrossRef]
- Innamorato, N.G.; Rojo, A.I.; Garcıa-Yague, A.J.; Yamamoto, M.; Ceballos, M.L.d.; Cuadrado, A. The Transcription Factor Nrf2 Is a Therapeutic Target against Brain Inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef]
- Yadav, S.K.; Soin, D.; Ito, K.; Dhib-Jalbut, S. Insight into the mechanism of action of dimethyl fumarate in multiple sclerosis. J. Mol. Med. 2019, 97, 463–472. [Google Scholar] [CrossRef]
- Al-Jaderi, Z.; Maghazachi, A.A. Utilization of Dimethyl Fumarate and Related Molecules for Treatment of Multiple Sclerosis, Cancer, and Other Diseases. Front. Immunol. 2016, 7, 278. [Google Scholar] [CrossRef]
- Mills, E.A.; Ogrodnik, M.A.; Plave, A.; Mao-Draayer, Y. Emerging Understanding of the Mechanism of Action for Dimethyl Fumarate in the Treatment of Multiple Sclerosis. Front. Neurol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Parodi, B.; Rossi, S.; Morando, S.; Cordano, C.; Bragoni, A.; Motta, C.; Usai, C.; Wipke, B.T.; Scannevin, R.H.; Mancardi, G.L.; et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015, 130, 279–295. [Google Scholar] [CrossRef]
- Hoeferlin, G.F.; Menendez, D.M.; Krebs, O.K.; Capadona, J.R.; Shoffstall, A.J. Assessment of Thermal Damage from Robot-Drilled Craniotomy for Cranial Window Surgery in Mice. JoVE, 2022, in press.
- Shoffstall, A.J.; Paiz, J.E.; Miller, D.M.; Rial, G.M.; Willis, M.T.; Menendez, D.M.; Hostler, S.R.; Capadona, J.R. Potential for thermal damage to the blood-brain barrier during craniotomy: Implications for intracortical recording microelectrodes. J. Neural Eng. 2018, 15, 034001. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Sturgill, B.; Radhakrishna, R.; Thai, T.T.; Patnaik, S.S.; Capadona, J.R.; Pancrazio, J.J. Characterization of Active Electrode Yield for Intracortical Arrays: Awake versus Anesthesia. Micromachines 2022, 13, 480. [Google Scholar] [CrossRef] [PubMed]
- Usoro, J.O.; Dogra, K.; Abbott, J.R.; Radhakrishna, R.; Cogan, S.F.; Pancrazio, J.J.; Patnaik, S.S. Influence of Implantation Depth on the Performance of Intracortical Probe Recording Sites. Micromachines 2021, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- Stiller, A.M.; Usoro, J.; Frewin, C.L.; Danda, V.R.; Ecker, M.; Joshi-Imre, A.; Musselman, K.C.; Voit, W.; Modi, R.; Pancrazio, J.J.; et al. Chronic Intracortical Recording and Electrochemical Stability of Thiol-ene/Acrylate Shape Memory Polymer Electrode Arrays. Micromachines 2018, 9, 500. [Google Scholar] [CrossRef]
- Ereifej, E.S.; Smith, C.S.; Meade, S.M.; Chen, K.; Feng, H.; Capadona, J.R. The neuroinflammatory response to nanopatterning parallel grooves into the surface structure of intracortical microelectrodes. Adv. Funct. Mater. 2018, 28, 1704420. [Google Scholar] [CrossRef]
- Nguyen, J.K.; Jorfi, M.; Buchanan, K.L.; Park, D.J.; Foster, E.J.; Tyler, D.J.; Rowan, S.J.; Weder, C.; Capadona, J.R. Influence of resveratrol release on the tissue response to mechanically adaptive cortical implants. Acta Biomater. 2016, 29, 81–93. [Google Scholar] [CrossRef]
- Potter, K.A.; Simon, J.S.; Velagapudi, B.; Capadona, J.R. Reduction of autofluorescence at the microelectrode-cortical tissue interface improves antibody detection. J. Neurosci. Methods 2012, 203, 96–105. [Google Scholar] [CrossRef]
- Lindner, S.C.; Yu, M.; Capadona, J.R.; Shoffstall, A.J. A graphical user interface to assess the neuroinflammatory response to intracortical microelectrodes. J. Neurosci. Methods 2019, 317, 141–148. [Google Scholar] [CrossRef]
- Stringer, C.; Wang, T.; Michaelos, M.; Pachitariu, M. Cellpose: A generalist algorithm for cellular segmentation. Nat. Methods 2021, 18, 100–106. [Google Scholar] [CrossRef]
- Bedell, H.W.; Schaub, N.J.; Capadona, J.R.; Ereifej, E.S. Differential expression of genes involved in the acute innate immune response to intracortical microelectrodes. Acta Biomater. 2020, 102, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Ramhoj, L.; Axelstad, M.; Svingen, T. Validation of endogenous reference genes in rat cerebral cortex for RT-qPCR analyses in developmental toxicity studies. PeerJ 2019, 7, e7181. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Regan, B.; Ereifej, E.S.; Chan, E.R.; Capadona, J.R. Neuroinflammatory Gene Expression Analysis Reveals Pathways of Interest as Potential Targets to Improve the Recording Performance of Intracortical Microelectrodes. Cells 2022, 11, 2348. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Druschel, L.N.; Chan, E.R.; Capadona, J.R. Differential expression of genes involved in the chronic response to intracortical microelectrodes. Acta Biomater. 2023, 169, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Readnower, R.D.; Chavko, M.; Adeeb, S.; Conroy, M.D.; Pauly, J.R.; McCarron, R.M.; Sullivan, P.G. Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J. Neurosci. Res. 2010, 88, 3530–3539. [Google Scholar] [CrossRef]
- McConnell, G.C.; Rees, H.D.; Levey, A.I.; Gutekunst, C.A.; Gross, R.E.; Bellamkonda, R.V. Implanted neural electrodes cause chronic, local inflammation that is correlated with local neurodegeneration. J. Neural Eng. 2009, 6, 056003. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Kim, C.C.; Ryba, B.E.; Niemi, E.C.; Bando, J.K.; Locksley, R.M.; Liu, J.; Nakamura, M.C.; Seaman, W.E. Traumatic brain injury induces macrophage subsets in the brain. Eur. J. Immunol. 2013, 43, 2010–2022. [Google Scholar] [CrossRef]
- Kumar, A.; Alvarez-Croda, D.M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. Microglial/Macrophage Polarization Dynamics following Traumatic Brain Injury. J. Neurotrauma 2016, 33, 1732–1750. [Google Scholar] [CrossRef]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular infiltration in traumatic brain injury. J. Neuroinflammation 2020, 17, 328. [Google Scholar] [CrossRef]
- Takmakov, P.; Ruda, K.; Phillips, K.S.; Isayeva, I.S.; Krauthamer, V.; Welle, C.G. Rapid evaluation of the durability of cortical neural implants using accelerated aging with reactive oxygen species. J. Neural Eng. 2015, 12, 026003. [Google Scholar] [CrossRef]
- Nolta, N.F.; Christensen, M.B.; Crane, P.D.; Skousen, J.L.; Tresco, P.A. BBB leakage, astrogliosis, and tissue loss correlate with silicon microelectrode array recording performance. Biomaterials 2015, 53, 753–762. [Google Scholar] [CrossRef]
- Saxena, T.; Karumbaiah, L.; Gaupp, E.A.; Patkar, R.; Patil, K.; Betancur, M.; Stanley, G.B.; Bellamkonda, R.V. The impact of chronic blood–brain barrier breach on intracortical electrode function. Biomaterials 2013, 34, 4703–4713. [Google Scholar] [CrossRef] [PubMed]
- Bonow, R.H.; Aid, S.; Zhang, Y.; Becker, K.G.; Bosetti, F. The brain expression of genes involved in inflammatory response, the ribosome, and learning and memory is altered by centrally injected lipopolysaccharide in mice. Pharmacogenomics J. 2009, 9, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Landsiedel, R.; Hahn, D.; Ossig, R.; Ritz, S.; Sauer, L.; Buesen, R.; Rehm, S.; Wohlleben, W.; Groeters, S.; Strauss, V.; et al. Gut microbiome and plasma metabolome changes in rats after oral gavage of nanoparticles: Sensitive indicators of possible adverse health effects. Part. Fibre Toxicol. 2022, 19, 1828. [Google Scholar] [CrossRef]
- Castaneda, M.; Smith, K.M.; Nixon, J.C.; Hernandez, C.J.; Rowan, S. Alterations to the gut microbiome impair bone tissue strength in aged mice. Bone Rep. 2021, 14, 101065. [Google Scholar] [CrossRef]
- Galland, L. The gut microbiome and the brain. J. Med. Food 2014, 17, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Jiang, Y.; Xu, K.; Cui, M.; Ye, W.; Zhao, G.; Jin, L.; Chen, X. The progress of gut microbiome research related to brain disorders. J. Neuroinflammation 2020, 17, 25. [Google Scholar] [CrossRef]
- Gershon, M.D.; Margolis, K.G. The gut, its microbiome, and the brain: Connections and communications. J. Clin. Investig. 2021, 131, e143768. [Google Scholar] [CrossRef]
- Hanscom, M.; Loane, D.J.; Shea-Donohue, T. Brain-gut axis dysfunction in the pathogenesis of traumatic brain injury. J. Clin. Investig. 2021, 131, e143777. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.G.; Cryan, J.F. Brain-Gut-Microbiota Axis and Mental Health. Psychosom. Med. 2017, 79, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Fulling, C.; Dinan, T.G.; Cryan, J.F. Gut Microbe to Brain Signaling: What Happens in Vagus. Neuron 2019, 101, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.P.; Dinger, N.; Levine, B.S. Stress produced by gavage administration in the rat. Contemp. Top. Lab. Anim. Sci. 2000, 39, 17–21. [Google Scholar] [PubMed]
- Dobrakovova, M.; Jurcovicova, J. Corticosterone and prolactin responses to repeated handling and transfer of male rats. Exp. Clin. Endocrinol. 1984, 83, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Zaro, B.W.; Vinogradova, E.V.; Lazar, D.C.; Blewett, M.M.; Suciu, R.M.; Takaya, J.; Studer, S.; de la Torre, J.C.; Casanova, J.L.; Cravatt, B.F.; et al. Dimethyl Fumarate Disrupts Human Innate Immune Signaling by Targeting the IRAK4-MyD88 Complex. J. Immunol. 2019, 202, 2737–2746. [Google Scholar] [CrossRef]
- Montes Diaz, G.; Fraussen, J.; Van Wijmeersch, B.; Hupperts, R.; Somers, V. Dimethyl fumarate induces a persistent change in the composition of the innate and adaptive immune system in multiple sclerosis patients. Sci. Rep. 2018, 8, 8194. [Google Scholar] [CrossRef]
- Winkelmann, A.; Loebermann, M.; Reisinger, E.C.; Hartung, H.P.; Zettl, U.K. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 217–233. [Google Scholar] [CrossRef]
- Longbrake, E.E.; Naismith, R.T.; Parks, B.J.; Wu, G.F.; Cross, A.H. Dimethyl fumarate-associated lymphopenia: Risk factors and clinical significance. Mult. Scler. J. Exp. Transl. Clin. 2015, 1, 2055217315596994. [Google Scholar] [CrossRef]
- Dobariya, A.; El Ahmadieh, T.Y.; Good, L.B.; Hernandez-Reynoso, A.G.; Jakkamsetti, V.; Brown, R.; Dunbar, M.; Ding, K.; Luna, J.; Kallem, R.R.; et al. Recording of pig neuronal activity in the comparative context of the awake human brain. Sci. Rep. 2022, 12, 15503. [Google Scholar] [CrossRef] [PubMed]
- Raspopovic, S.; Cimolato, A.; Panarese, A.; Vallone, F.; Del Valle, J.; Micera, S.; Navarro, X. Neural signal recording and processing in somatic neuroprosthetic applications. A review. J. Neurosci. Methods 2020, 337, 108653. [Google Scholar] [CrossRef] [PubMed]
- Carnicer-Lombarte, A.; Chen, S.T.; Malliaras, G.G.; Barone, D.G. Foreign Body Reaction to Implanted Biomaterials and Its Impact in Nerve Neuroprosthetics. Front. Bioeng. Biotechnol. 2021, 9, 622524. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Custom Gene Panel | NanoString Gene Panel | Housekeeping Gene Panel | ||||||
|---|---|---|---|---|---|---|---|---|
| AIM2 | Ercc6 | Nr2f6 | Abl1 | Cycs | Hspb1 | Nos1 | Tnf | Hprt |
| ARC | FCER1G | Osgin1 | Ager | Ddit3 | Htra2 | Nos3 | Tor1a | Rpl13a |
| Bdnf | FCGR2B | OSMR | Aif1 | Dnm2 | Idh1 | Nr4a2 | Tpm1 | Rps18 |
| BLNK | GFAP | Prnp | Akt1 | Ep300 | Il1r1 | Oxr1 | Trp53 | Sdha |
| C3 | GSTA1 | PSMB8 | Apoe | Fas | Il6 | Park7 | Trpm2 | Tbp |
| C3AR1 | Gsta2 | Ptgs2 | App | Fn1 | Ins2 | Parp1 | Txnl1 | Ubc |
| C4A | GSTM2 | PTPN6 | Atf4 | Fos | Ipcef1 | Pdgfrb | Ubqln1 | |
| C5AR1 | Hmox1 | PTX3 | Atp13a2 | Fxn | Jun | Pink1 | Xbp1 | |
| CASP8 | Il1b | SCD1 | Atp7a | Gnao1 | Lpo | Pla2g4a | ||
| CCL1 | IL2RG | SERPINA3N | Atrn | Gpr37 | Lrrk2 | Ppargc1a | ||
| CD14 | IRAK4 | Sod3 | Bad | Gsk3b | Mapt | Psen1 | ||
| CD36 | IRF7 | SPP1 | Bcl2 | Gsr | Mgmt | Rela | ||
| CD45 | ITGAM | Srxn1 | Bnip3 | Gss | Mmp14 | Sirt1 | ||
| CD68 | KEAP1 | TNFRSF1A | Casp3 | Gstp1 | Mutyh | Sirt2 | ||
| CD74 | LILRB4A | TNFRSF25 | Ccl5 | Gucy1b3 | Ncf1 | Slc8a1 | ||
| CD84 | MMP12 | Txnrd1 | Ccs | H2-T23 | Nefh | Snca | ||
| CLEC7A | MPEG1 | TYROBP | Cdk2 | Hdac2 | Ngfg | Sod1 | ||
| CTSS | Nfe2l2 | Vegfa | CIM | Hdac6 | Ngfr | Sod2 | ||
| DOCK2 | Noxa1 | Cln8 | Hgf | Nme5 | Src | |||
| Ehd2 | Nqo1 | Cybb | Hif1a | Nol3 | Stx2 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoeferlin, G.F.; Bajwa, T.; Olivares, H.; Zhang, J.; Druschel, L.N.; Sturgill, B.S.; Sobota, M.; Boucher, P.; Duncan, J.; Hernandez-Reynoso, A.G.; et al. Antioxidant Dimethyl Fumarate Temporarily but Not Chronically Improves Intracortical Microelectrode Performance. Micromachines 2023, 14, 1902. https://doi.org/10.3390/mi14101902
Hoeferlin GF, Bajwa T, Olivares H, Zhang J, Druschel LN, Sturgill BS, Sobota M, Boucher P, Duncan J, Hernandez-Reynoso AG, et al. Antioxidant Dimethyl Fumarate Temporarily but Not Chronically Improves Intracortical Microelectrode Performance. Micromachines. 2023; 14(10):1902. https://doi.org/10.3390/mi14101902
Chicago/Turabian StyleHoeferlin, George F., Tejas Bajwa, Hannah Olivares, Jichu Zhang, Lindsey N. Druschel, Brandon S. Sturgill, Michael Sobota, Pierce Boucher, Jonathan Duncan, Ana G. Hernandez-Reynoso, and et al. 2023. "Antioxidant Dimethyl Fumarate Temporarily but Not Chronically Improves Intracortical Microelectrode Performance" Micromachines 14, no. 10: 1902. https://doi.org/10.3390/mi14101902
APA StyleHoeferlin, G. F., Bajwa, T., Olivares, H., Zhang, J., Druschel, L. N., Sturgill, B. S., Sobota, M., Boucher, P., Duncan, J., Hernandez-Reynoso, A. G., Cogan, S. F., Pancrazio, J. J., & Capadona, J. R. (2023). Antioxidant Dimethyl Fumarate Temporarily but Not Chronically Improves Intracortical Microelectrode Performance. Micromachines, 14(10), 1902. https://doi.org/10.3390/mi14101902