Interfacing Digital Microfluidics with Ambient Mass Spectrometry Using SU-8 as Dielectric Layer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
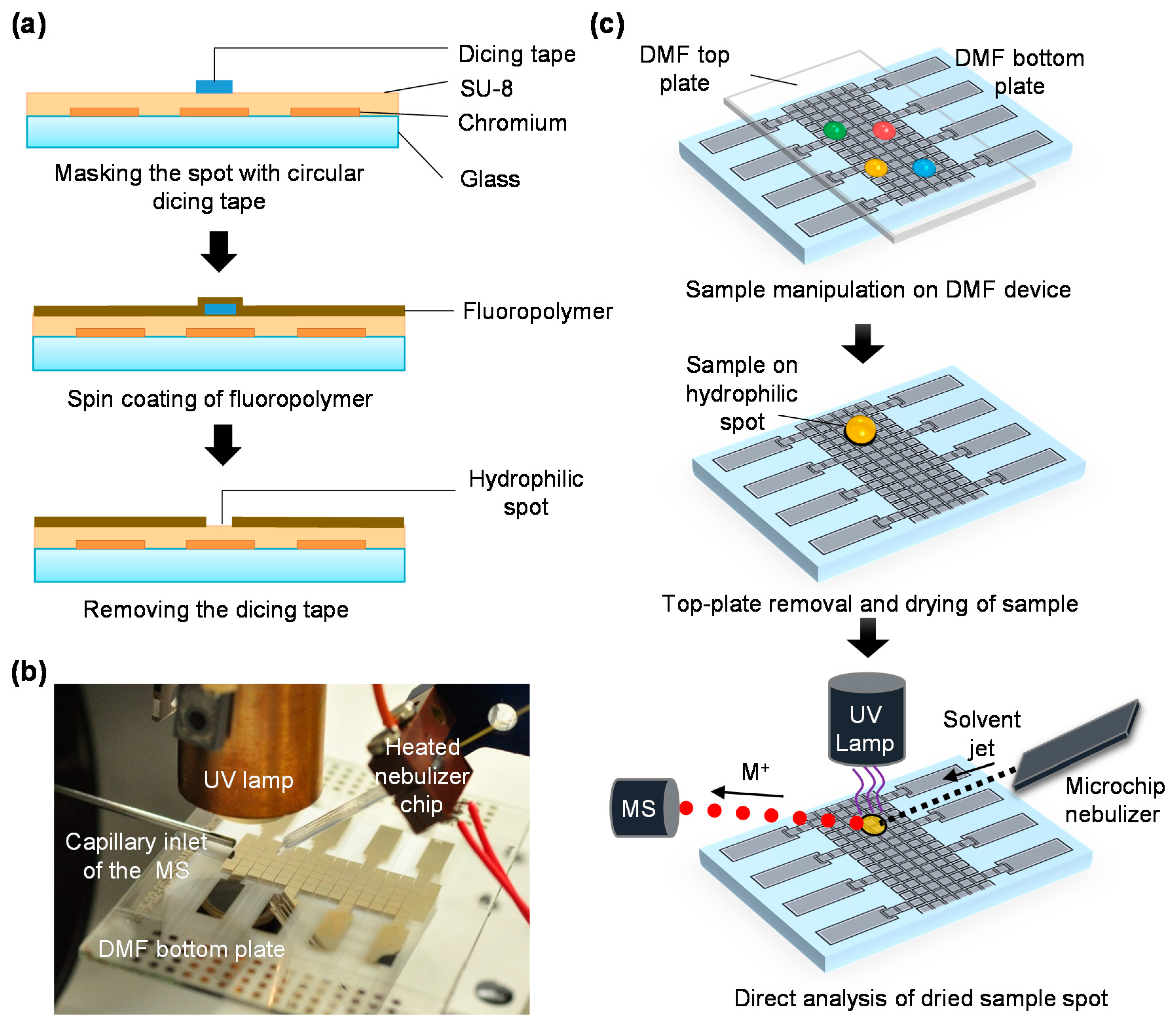
2.2. Digital Microfluidic Chip Fabrication
2.3. Mass Spectrometry
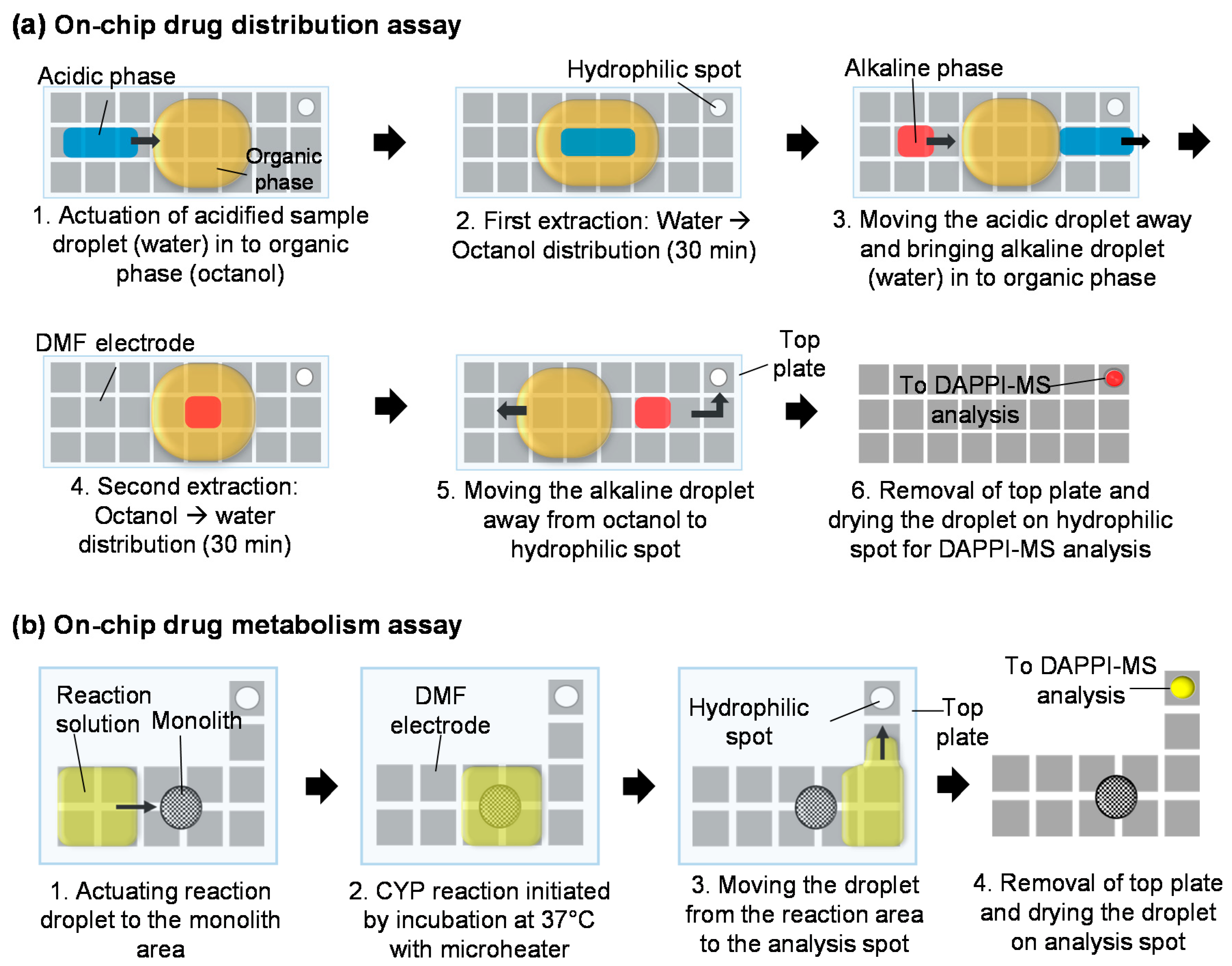
2.4. On-Chip Drug Distribution Assays
2.5. Preparation of Immobilized Cytochrome P450 Reactors
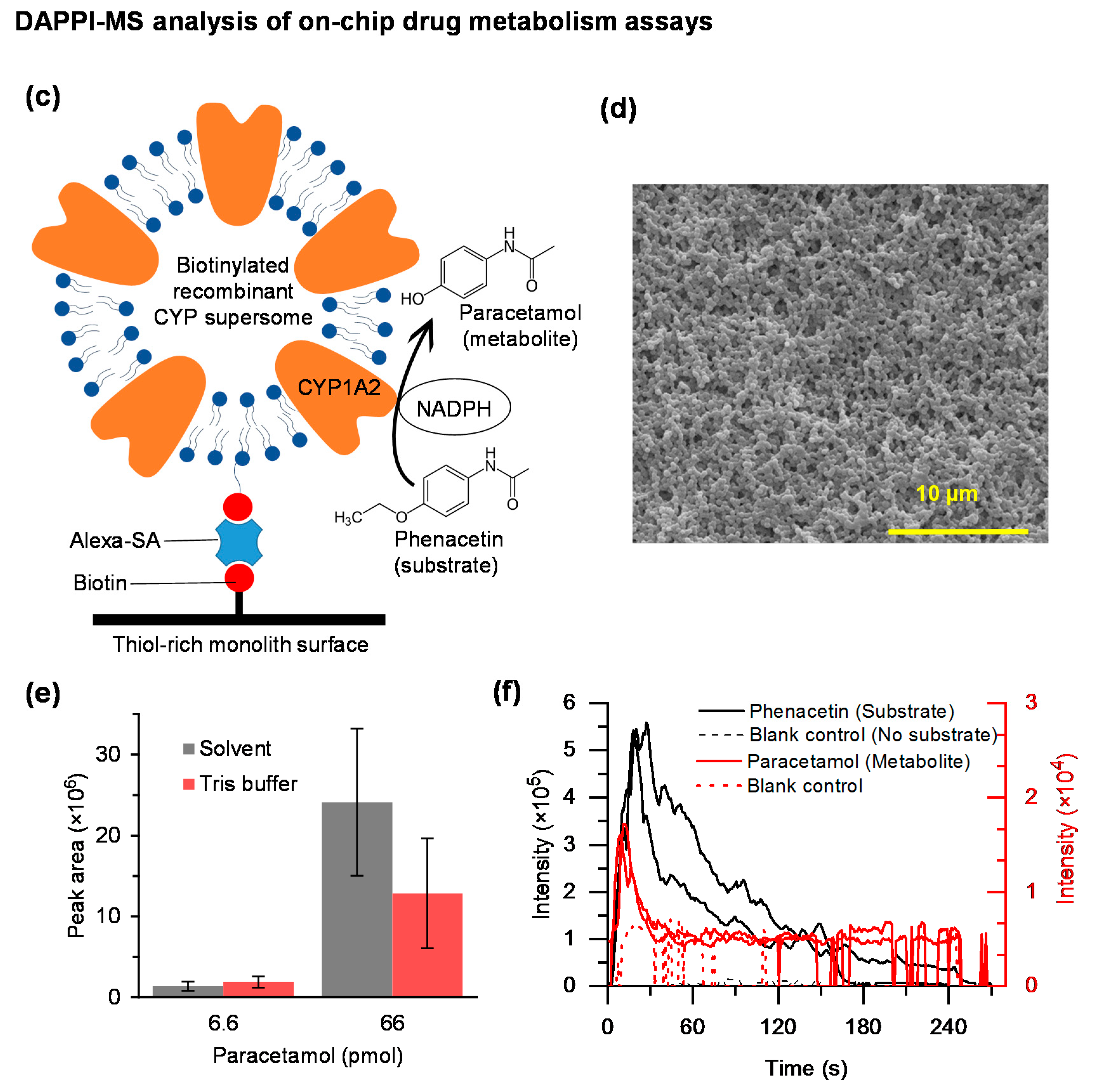
2.6. On-Chip Drug Metabolism Assays
3. Results and Discussion
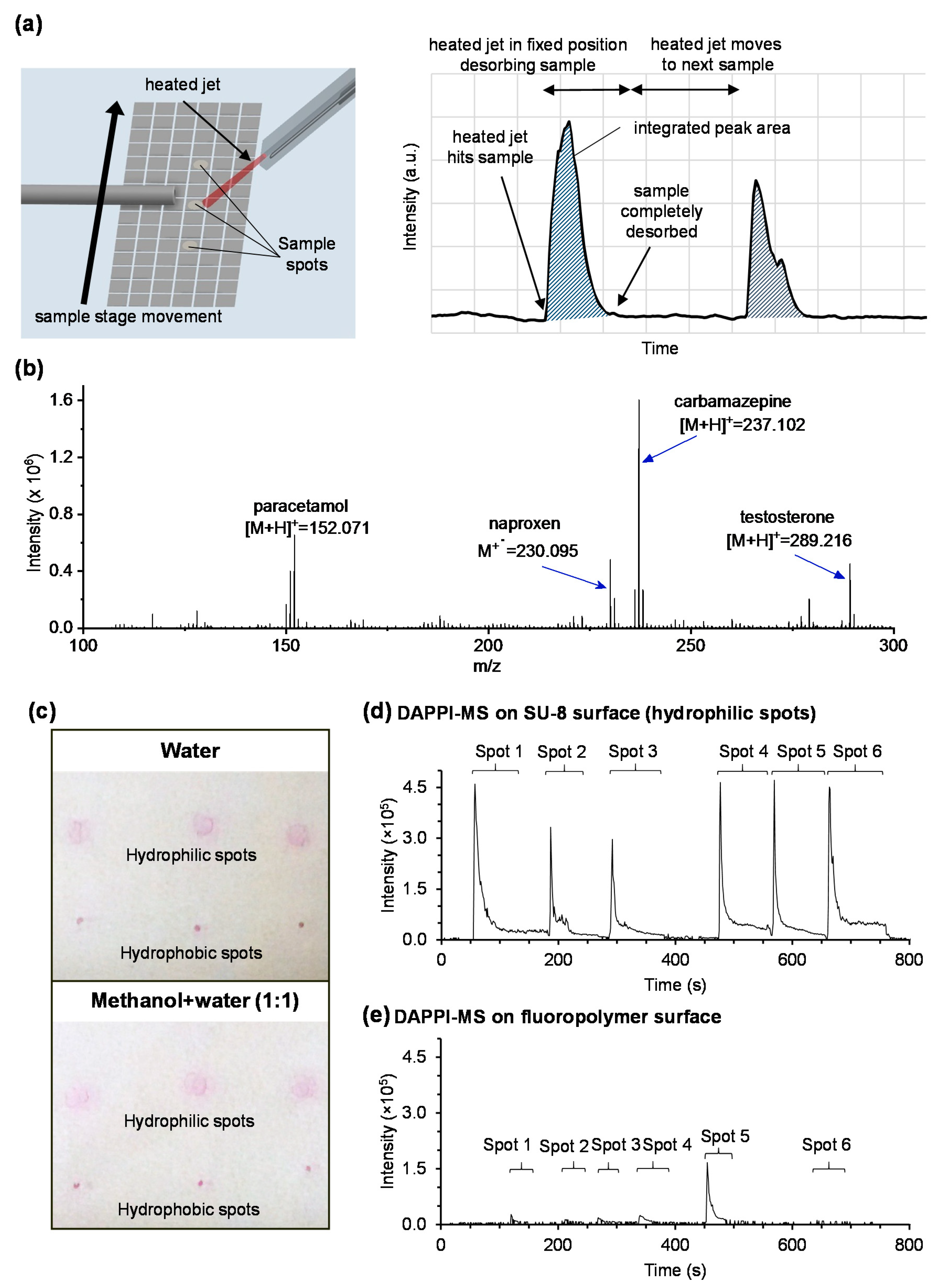
3.1. Material Considerations
3.2. Mass Spectrometry Method Qualification
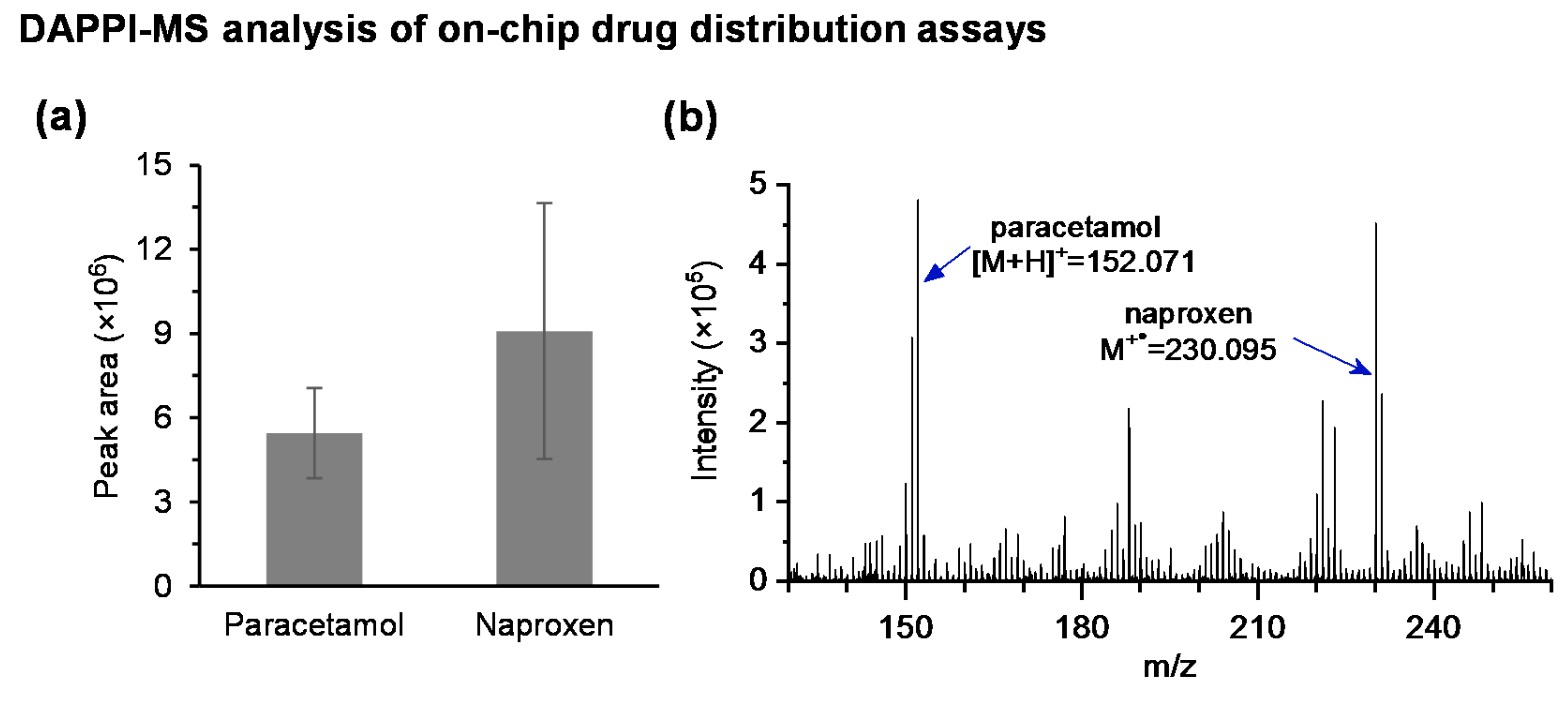
3.3. On-Chip DAPPI-MS Analysis of Drug Distribution Assays
3.4. On-Chip DAPPI-MS Analysis of Drug Metabolism Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Choi, K.; Ng, A.H.C.; Fobel, R.; Wheeler, A.R. Digital Microfluidics. Annu. Rev. Anal. Chem. 2012, 5, 413–440. [Google Scholar] [CrossRef] [PubMed]
- Pollack, M.G.; Shenderov, A.D.; Fair, R.B. Electrowetting-based actuation of droplets for integrated microfluidics. Lab Chip 2002, 2, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.K.; Moon, H.; Kim, C.-J. Creating, transporting, cutting, and merging liquid droplets by electrowetting-based actuation for digital microfluidic circuits. J. Microelectromech. Syst. 2003, 12, 70–80. [Google Scholar] [Green Version]
- Liu, H.; Dharmatilleke, S.; Maurya, D.K.; Tay, A.A.O. Dielectric materials for electrowetting-on-dielectric actuation. Microsyst. Technol. 2009, 16, 449–460. [Google Scholar] [CrossRef]
- Kahouli, A.; Sylvestre, A.; Ortega, L.; Jomni, F.; Yangui, B.; Maillard, M.; Berge, B.; Robert, J.-C.; Legrand, J. Structural and dielectric study of parylene C thin films. Appl. Phys. Lett. 2009, 94, 152901. [Google Scholar] [CrossRef]
- Choy, K.L. Chemical vapour deposition of coatings. Prog. Mater. Sci. 2003, 48, 57–170. [Google Scholar] [CrossRef]
- Sathyanarayanan, G.; Haapala, M.; Kiiski, I.; Sikanen, T. Digital microfluidic immobilized cytochrome P450 reactors with integrated inkjet-printed microheaters for droplet-based drug metabolism research. Anal. Bioanal. Chem. 2018, 410, 6677–6687. [Google Scholar] [CrossRef]
- Arscott, S. SU-8 as a material for lab-on-a-chip-based mass spectrometry. Lab Chip 2014, 14, 3668–3689. [Google Scholar] [CrossRef]
- Sikanen, T.; Heikkilä, L.; Tuomikoski, S.; Ketola, R.A.; Kostiainen, R.; Franssila, S.; Kotiaho, T. Performance of SU-8 Microchips as Separation Devices and Comparison with Glass Microchips. Anal. Chem. 2007, 79, 6255–6263. [Google Scholar] [CrossRef]
- Nissilä, T.; Sainiemi, L.; Franssila, S.; Ketola, R.A. Fully polymeric integrated microreactor/electrospray ionization chip for on-chip digestion and mass spectrometric analysis. Sens. Actuators B Chem. 2009, 143, 414–420. [Google Scholar] [CrossRef]
- Becker, H.; Gärtner, C. Polymer microfabrication technologies for microfluidic systems. Anal. Bioanal. Chem. 2008, 390, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Dryden, M.D.M.; Rackus, D.D.G.; Shamsi, M.H.; Wheeler, A.R. Integrated Digital Microfluidic Platform for Voltammetric Analysis. Anal. Chem. 2013, 85, 8809–8816. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Jia, Y.; Dong, C.; Gao, J.; Mak, P.-I.; Martins, R.P. Sub-7-second genotyping of single-nucleotide polymorphism by high-resolution melting curve analysis on a thermal digital microfluidic device. Lab Chip 2016, 16, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, H.; Despont, M.; Fahrni, N.; LaBianca, N.; Renaud, P.; Vettiger, P. SU-8: A low-cost negative resist for MEMS. J. Micromech. Microeng. 1997, 7, 121. [Google Scholar] [CrossRef]
- Eberlin, L.S.; Ferreira, C.R.; Dill, A.L.; Ifa, D.R.; Cheng, L.; Cooks, R.G. Non-Destructive, Histologically Compatible Tissue Imaging by Desorption Electrospray Ionization Mass Spectrometry. Chembiochem. Eur. J. Chem. Biol. 2011, 12, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.U.; Tata, A.; Wu, C.; Perry, R.H.; Haas, G.; West, L.; Graham Cooks, R. Direct analysis of Stevia leaves for diterpene glycosides by desorption electrospray ionization mass spectrometry. Analyst 2009, 134, 867–874. [Google Scholar] [CrossRef]
- Morelato, M.; Beavis, A.; Kirkbride, P.; Roux, C. Forensic applications of desorption electrospray ionisation mass spectrometry (DESI-MS). Forensic Sci. Int. 2013, 226, 10–21. [Google Scholar] [CrossRef]
- Pavlovich, M.J.; Musselman, B.; Hall, A.B. Direct analysis in real time—Mass spectrometry (DART-MS) in forensic and security applications. Mass Spectrom. Rev. 2018, 37, 171–187. [Google Scholar] [CrossRef]
- Shih, S.C.C.; Yang, H.; Jebrail, M.J.; Fobel, R.; McIntosh, N.; Al-Dirbashi, O.Y.; Chakraborty, P.; Wheeler, A.R. Dried Blood Spot Analysis by Digital Microfluidics Coupled to Nanoelectrospray Ionization Mass Spectrometry. Anal. Chem. 2012, 84, 3731–3738. [Google Scholar] [CrossRef]
- Kirby, A.E.; Wheeler, A.R. Microfluidic origami: A new device format for in-line reaction monitoring by nanoelectrospray ionization mass spectrometry. Lab Chip 2013, 13, 2533–2540. [Google Scholar] [CrossRef]
- Choi, K.; Boyacı, E.; Kim, J.; Seale, B.; Barrera-Arbelaez, L.; Pawliszyn, J.; Wheeler, A.R. A digital microfluidic interface between solid-phase microextraction and liquid chromatography–mass spectrometry. J. Chromatogr. A 2016, 1444, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Wheeler, A.R.; Garrell, R.L.; Loo, J.A.; Kim, C.-J. An integrated digital microfluidic chip for multiplexed proteomic sample preparation and analysis by MALDI-MS. Lab Chip 2006, 6, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Nichols, K.P.; Gardeniers, J.G.E. A Digital Microfluidic System for the Investigation of Pre-Steady-State Enzyme Kinetics Using Rapid Quenching with MALDI-TOF Mass Spectrometry. Anal. Chem. 2007, 79, 8699–8704. [Google Scholar] [CrossRef] [PubMed]
- Haapala, M.; Pól, J.; Saarela, V.; Arvola, V.; Kotiaho, T.; Ketola, R.A.; Franssila, S.; Kauppila, T.J.; Kostiainen, R. Desorption Atmospheric Pressure Photoionization. Anal. Chem. 2007, 79, 7867–7872. [Google Scholar] [CrossRef] [PubMed]
- Suni, N.M.; Lindfors, P.; Laine, O.; Östman, P.; Ojanperä, I.; Kotiaho, T.; Kauppila, T.J.; Kostiainen, R. Matrix effect in the analysis of drugs of abuse from urine with desorption atmospheric pressure photoionization-mass spectrometry (DAPPI-MS) and desorption electrospray ionization-mass spectrometry (DESI-MS). Anal. Chim. Acta 2011, 699, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Luosujärvi, L.; Arvola, V.; Haapala, M.; Pól, J.; Saarela, V.; Franssila, S.; Kotiaho, T.; Kostiainen, R.; Kauppila, T.J. Desorption and Ionization Mechanisms in Desorption Atmospheric Pressure Photoionization. Anal. Chem. 2008, 80, 7460–7466. [Google Scholar] [CrossRef]
- Fobel, R.; Fobel, C.; Wheeler, A.R. DropBot: An open-source digital microfluidic control system with precise control of electrostatic driving force and instantaneous drop velocity measurement. Appl. Phys. Lett. 2013, 102, 193513. [Google Scholar] [CrossRef]
- Saarela, V.; Haapala, M.; Kostiainen, R.; Kotiaho, T.; Franssila, S. Glass microfabricated nebulizer chip for mass spectrometry. Lab Chip 2007, 7, 644–646. [Google Scholar] [CrossRef]
- Feidenhans’l, N.A.; Lafleur, J.P.; Jensen, T.G.; Kutter, J.P. Surface functionalized thiol-ene waveguides for fluorescence biosensing in microfluidic devices. Electrophoresis 2014, 35, 282–288. [Google Scholar] [CrossRef]
- Kiiski, I.; Pihlaja, T.; Urvas, L.; Wiedmer, S.; Jokinen, V.; Sikanen, T. Overcoming the Pitfalls of Cytochrome P450 Immobilization Through the Use of Fusogenic Liposomes. Adv. Biosyst. 2018, accepted. [Google Scholar] [CrossRef]
- Melai, J.; Salm, C.; Smits, S.; Visschers, J.; Schmitz, J. The electrical conduction and dielectric strength of SU-8. J. Micromech. Microeng. 2009, 19, 065012. [Google Scholar] [CrossRef]
- Fraunhofer Insititute for Silicate Research ISC. Available online: https://www.isc.fraunhofer.de/content/dam/isc/de/documents/Publikationen/Dielectric_ORMOCERs_for_system_in_package_electronics.pdf (accessed on 19 November 2018).
- Li, Y.-F.; Sheng, Y.-J.; Tsao, H.-K. Evaporation Stains: Suppressing the Coffee-Ring Effect by Contact Angle Hysteresis. Langmuir 2013, 29, 7802–7811. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Paracetamol | Naproxen | Carbamazepine | Testosterone |
| Molecular Formula | C8H9NO2 | C14H14O3 | C15H12N2O | C19H28O2 |
| Observed Ion Type | 152.071 ([M + H]+) | 230.095 (M+•) | 237.102 ([M + H]+) | 289.216 ([M + H]+) |
| Monoisotopic Mass (g/mol) | 151.063 | 230.094 | 236.095 | 288.209 |
| pKa (acidic) | 9.46 | 4.19 | 15.96 | n.d. (internal qualifier) |
| LogD (pH 2) LogD (pH 12) | 0.9 −1.15 | 2.98 −0.54 | 2.76 2.76 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sathyanarayanan, G.; Haapala, M.; Sikanen, T. Interfacing Digital Microfluidics with Ambient Mass Spectrometry Using SU-8 as Dielectric Layer. Micromachines 2018, 9, 649. https://doi.org/10.3390/mi9120649
Sathyanarayanan G, Haapala M, Sikanen T. Interfacing Digital Microfluidics with Ambient Mass Spectrometry Using SU-8 as Dielectric Layer. Micromachines. 2018; 9(12):649. https://doi.org/10.3390/mi9120649
Chicago/Turabian StyleSathyanarayanan, Gowtham, Markus Haapala, and Tiina Sikanen. 2018. "Interfacing Digital Microfluidics with Ambient Mass Spectrometry Using SU-8 as Dielectric Layer" Micromachines 9, no. 12: 649. https://doi.org/10.3390/mi9120649
APA StyleSathyanarayanan, G., Haapala, M., & Sikanen, T. (2018). Interfacing Digital Microfluidics with Ambient Mass Spectrometry Using SU-8 as Dielectric Layer. Micromachines, 9(12), 649. https://doi.org/10.3390/mi9120649