Targeting Epigenetic Aberrations in Pancreatic Cancer, a New Path to Improve Patient Outcomes?


Abstract
:1. Introduction
2. Targeting Epigenetic Pathways as a Way to Reprogram Tumor Biology
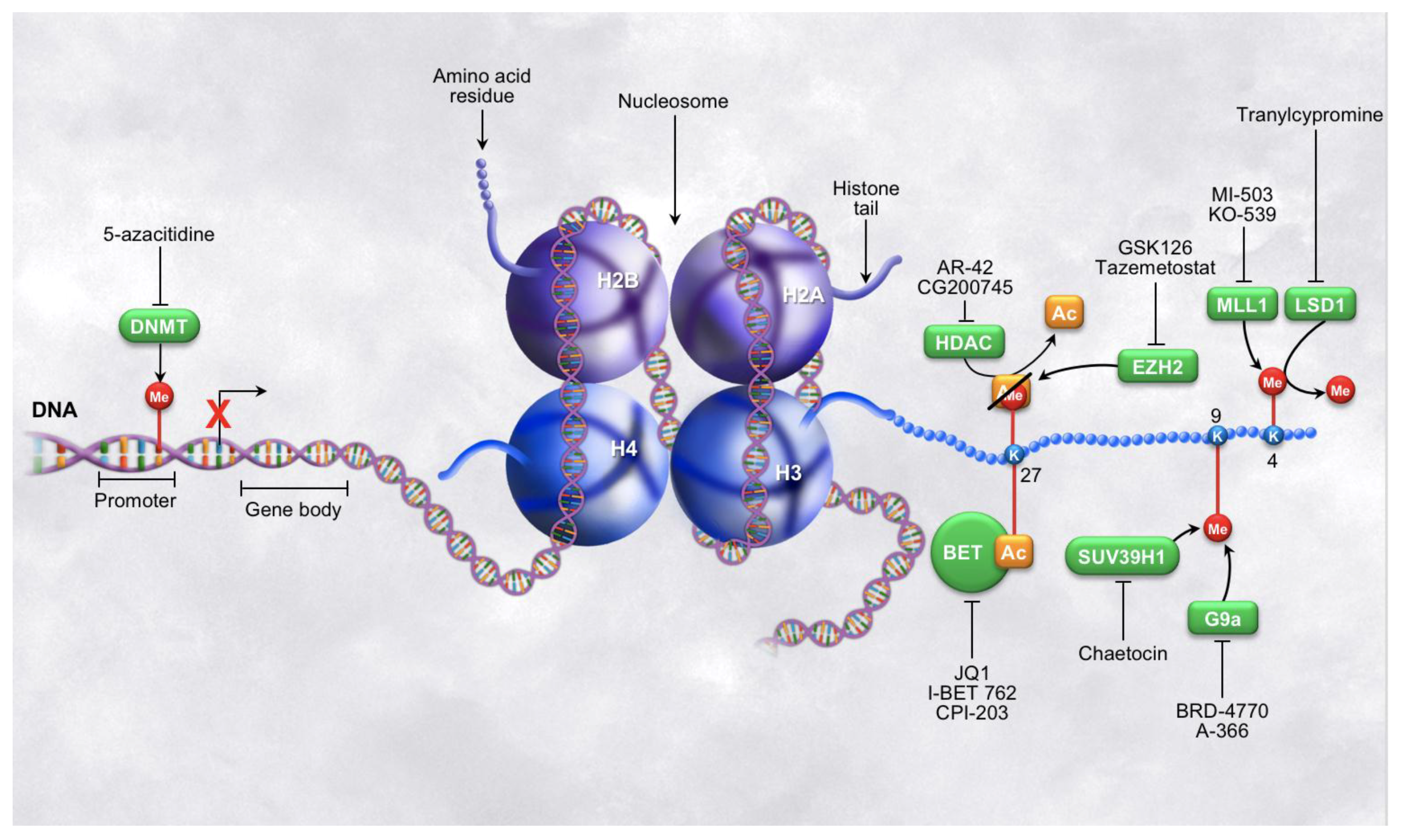
3. Targeting DNA Methylation
4. Targeting Histone Modifications
5. Challenges in the Field
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Society, A.C. Cancer Facts & Figures 2017; American Cancer Society: Atlanta, GA, USA, 2017. [Google Scholar]
- Johns Hopkins University. Types of Neoplasms of the Pancreas. Available online: http://pathology.jhu.edu/pc/BasicTypes2.php?area=ba (accessed on 10 November 2017).
- Pancreatic Cancer Facts 2017; Pancreatic Cancer Action Network: Manhattan Beach, CA, USA, 2017; p. 2.
- Garcia, M.N.; Grasso, D.; Lopez-Millan, M.B.; Hamidi, T.; Loncle, C.; Tomasini, R.; Lomberk, G.; Porteu, F.; Urrutia, R.; Iovanna, J.L. IER3 supports KRASG12D-dependent pancreatic cancer development by sustaining ERK1/2 phosphorylation. J. Clin. Investig. 2014, 124, 4709–4722. [Google Scholar] [CrossRef] [PubMed]
- McCleary-Wheeler, A.L.; Lomberk, G.A.; Weiss, F.U.; Schneider, G.; Fabbri, M.; Poshusta, T.L.; Dusetti, N.J.; Baumgart, S.; Iovanna, J.L.; Ellenrieder, V.; et al. Insights into the epigenetic mechanisms controlling pancreatic carcinogenesis. Cancer Lett. 2013, 328, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Silverman, B.R.; Shi, J. Alterations of epigenetic regulators in pancreatic cancer and their clinical implications. Int. J. Mol. Sci. 2016, 17, 2138. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Epigenetics 2016, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Sahin, F.; Iacobuzio-Donahue, C.A.; Garcia-Carracedo, D.; Wang, W.M.; Kuo, C.Y.; Chen, D.; Arking, D.E.; Lowy, A.M.; Hruban, R.H.; et al. Disruption of p16 and activation of kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget 2011, 2, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.M.; Kang, E.J.; Kang, K.; Kim, S.D.; Yang, K.; Yi, J.M. Combinatorial effects of an epigenetic inhibitor and ionizing radiation contribute to targeted elimination of pancreatic cancer stem cell. Oncotarget 2017, 8, 89005–89020. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Ray, A.; Van Brocklin, M.; Burnett, D.M.; Bowen, R.C.; Dyess, D.L.; Butler, T.W.; Dumlao, T.; Khong, H.T. A phase I trial of azacitidine and nanoparticle albumin bound paclitaxel in patients with advanced or metastatic solid tumors. Oncotarget 2017, 8, 52413–52419. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhou, Y.; Liu, X.; Shen, Y. GDC-0449 improves the antitumor activity of nano-doxorubicin in pancreatic cancer in a fibroblast-enriched microenvironment. Sci. Rep. 2017, 7, 13379. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.H.; Elliott, I.A.; Wu, N.; Matsumura, C.; Vogelauer, M.; Attar, N.; Dann, A.; Ghukasyan, R.; Toste, P.A.; Patel, S.G.; et al. Histone deacetylase inhibitors provoke a tumor supportive phenotype in pancreatic cancer associated fibroblasts. Oncotarget 2017, 8, 19074–19088. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Hoshikawa, M.; Tanaka, M.; Nakatsuka, T.; Fujiwara, H.; Miyabayashi, K.; Takahashi, R.; Tanaka, Y.; et al. Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget 2016, 7, 61469–61484. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Wang, J.; Jung, Y.; Cackowski, F.C.; Taichman, R.S. Reduction of histone marks, H3k9me3 and H3k27me3 by epidrug induces neuroendocrine differentiation in prostate cancer. J. Cell. Biochem. 2018, 119, 3697–3705. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, A.; Tsukada, Y.-I. DNA and Histone Methylation as Cancer Targets; Springer: Berlin, Germany, 2017; p. 624. [Google Scholar]
- Huang, S.; Litt, M.; Blakey, C.A. (Eds.) Epigenetic Gene Expression and Regulation; Epigenetic Gene Expression and Regulation; Academic Press: Cambridge, MA, USA, 2015; p. 482. [Google Scholar]
- Neureiter, D.; Jager, T.; Ocker, M.; Kiesslich, T. Epigenetics and pancreatic cancer: Pathophysiology and novel treatment aspects. World J. Gastroenterol. 2014, 20, 7830–7848. [Google Scholar] [CrossRef] [PubMed]
- Jakel, C.; Bergmann, F.; Toth, R.; Assenov, Y.; van der Duin, D.; Strobel, O.; Hank, T.; Kloppel, G.; Dorrell, C.; Grompe, M.; et al. Genome-wide genetic and epigenetic analyses of pancreatic acinar cell carcinomas reveal aberrations in genome stability. Nat. Commun. 2017, 8, 1323. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, S.D.; Madsen, P.H.; Larsen, A.C.; Johansen, M.B.; Pedersen, I.S.; Krarup, H.; Thorlacius-Ussing, O. Promoter hypermethylation in plasma-derived cell-free DNA as a prognostic marker for pancreatic adenocarcinoma staging. Int. J. Cancer 2017, 141, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Vakoc, C.R.; Tuveson, D.A. Untangling the genetics from the epigenetics in pancreatic cancer metastasis. Nat. Genet. 2017, 49, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xia, W.; Zhang, Z.; Liu, J.; Wang, H.; Adsay, N.V.; Albarracin, C.; Yu, D.; Abbruzzese, J.L.; Mills, G.B.; et al. Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol. Carcinog. 2008, 47, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Piccolo, S.R.; Cheng, L.; Soldi, R.; Han, B.; Johnson, W.E.; Bild, A.H. Genomic pathway analysis reveals that EZH2 and HDAC4 represent mutually exclusive epigenetic pathways across human cancers. BMC Med. Genom. 2013, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Yamauchi, N.; Shibahara, J.; Kimura, H.; Morikawa, T.; Ishikawa, S.; Nagae, G.; Nishi, A.; Sakamoto, Y.; Kokudo, N.; et al. Concurrent activation of acetylation and tri-methylation of H3K27 in a subset of hepatocellular carcinoma with aggressive behavior. PLoS ONE 2014, 9, e91330. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Paschall, A.V.; Shi, H.; Savage, N.; Waller, J.L.; Sabbatini, M.E.; Oberlies, N.H.; Pearce, C.; Liu, K. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Watanabe, H.; Peng, S.; Francis, J.M.; Kaplan, N.; Pedamallu, C.S.; Ramachandran, A.; Agoston, A.; Bass, A.J.; Meyerson, M. Dynamic epigenetic regulation by menin during pancreatic islet tumor formation. Mol. Cancer Res. MCR 2015, 13, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Hu, H.; Yuan, C.; Jin, Z.; Guo, Z.; Wang, L.; Wang, L. Histone deacetylase 3 promotes pancreatic cancer cell proliferation, invasion and increases drug-resistance through histone modification of P27, P53 and Bax. Int. J. Oncol. 2014, 45, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nahar, S.; Nakagawa, A.; Fernandez-Barrena, M.G.; Mertz, J.A.; Bryant, B.M.; Adams, C.E.; Mino-Kenudson, M.; Von Alt, K.N.; Chang, K.; et al. Regulation of GLI underlies a role for BET bromodomains in pancreatic cancer growth and the tumor microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4259–4270. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, R.J.; Gatignol, A. RNA interference therapies for an HIV-1 functional cure. Viruses 2017, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Feng, L.; Wu, W.; Weng, T.; Hu, C.; Hong, B.; Wang, F.X.C.; Shen, L.; Wang, Q.; Jin, X.; et al. MicroRNA expression profiling of pancreatic cancer cell line L3.6p1 following B7-H4 knockdown. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 44, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Drury, R.E.; O’Connor, D.; Pollard, A.J. The clinical application of microRNAs in infectious disease. Front. Immunol. 2017, 8, 1182. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.W.; Cho, W.C. The emerging role of miRNAs in combined cancer therapy. Expert Opin. Biol. Ther. 2015, 15, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Okada, F.; Ochiya, T. MiRNA therapy targeting cancer stem cells: A new paradigm for cancer treatment and prevention of tumor recurrence. Ther. Deliv. 2015, 6, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Gomes, K.M.; Costa, I.C.; Santos, J.F.; Dourado, P.M.; Forni, M.F.; Ferreira, J.C. Induced pluripotent stem cells reprogramming: Epigenetics and applications in the regenerative medicine. Rev. Assoc. Med. Bras. (1992) 2017, 63, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Kochat, V.; Equbal, Z.; Baligar, P.; Kumar, V.; Srivastava, M.; Mukhopadhyay, A. JMJD3 aids in reprogramming of bone marrow progenitor cells to hepatic phenotype through epigenetic activation of hepatic transcription factors. PLoS ONE 2017, 12, e0173977. [Google Scholar] [CrossRef] [PubMed]
- Tucker, D.W.; Getchell, C.R.; McCarthy, E.T.; Ohman, A.W.; Sasamoto, N.; Xu, S.; Ko, J.Y.; Gupta, M.; Shafrir, A.L.; Medina, J.E.; et al. Epigenetic reprogramming strategies to reverse global loss of 5-hydroxymethylcytosine, a prognostic factor for poor survival in high-grade serous ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 24, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Hah, N.; Littlewood, T.D.; Sodir, N.M.; Campos, T.; Downes, M.; Evans, R.M. Re-engineering the pancreas tumor microenvironment: A “regenerative program” hacked. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.L.; Mann, D.A.; Borthwick, L.A. Epigenetic reprogramming in liver fibrosis and cancer. Adv. Drug Deliv. Rev. 2017, 121, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Bhuvanalakshmi, G.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A.; Warrier, S. Epigenetic reprogramming converts human wharton’s jelly mesenchymal stem cells into functional cardiomyocytes by differential regulation of wnt mediators. Stem Cell Res. Ther. 2017, 8, 185. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Tsai, H.C.; Yen, R.C.; Zhang, Y.W.; Kong, X.; Wang, W.; Xia, L.; Baylin, S.B. Critical threshold levels of DNA methyltransferase 1 are required to maintain DNA methylation across the genome in human cancer cells. Genome Res. 2017, 27, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Abdelfatah, E.; Kerner, Z.; Nanda, N.; Ahuja, N. Epigenetic therapy in gastrointestinal cancer: The right combination. Ther. Adv. Gastroenterol. 2016, 9, 560–579. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Shukla, S.; Lakra, A.D.; Meeran, S.M.; Siddiqi, M.I. Identification of potent inhibitors of DNA methyltransferase 1 (DNMT1) through a pharmacophore-based virtual screening approach. J. Mol. Graph. Model. 2017, 75, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Rondelet, G.; Fleury, L.; Faux, C.; Masson, V.; Dubois, J.; Arimondo, P.B.; Willems, L.; Wouters, J. Inhibition studies of DNA methyltransferases by maleimide derivatives of RG108 as non-nucleoside inhibitors. Future Med. Chem. 2017, 9, 1465–1481. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Yin, W.J.; Lu, J.S.; Wang, L.; Wu, J.; Wu, F.Y.; Di, G.H.; Shen, Z.Z.; Shao, Z.M. ERα negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J. Cancer Res. Clin. Oncol. 2008, 134, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Gore, S.D.; Baylin, S.; Sugar, E.; Carraway, H.; Miller, C.B.; Carducci, M.; Grever, M.; Galm, O.; Dauses, T.; Karp, J.E.; et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006, 66, 6361–6369. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Cesaroni, M.; Chung, W.; Panjarian, S.; Tran, A.; Madzo, J.; Okamoto, Y.; Zhang, H.; Chen, X.; Jelinek, J.; et al. Transcriptional selectivity of epigenetic therapy in cancer. Cancer Res. 2017, 77, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.R.; Hsu, M.C.; Chen, L.T.; Hung, W.C. G9a orchestrates PCL3 and KDM7A to promote histone H3K27 methylation. Sci. Rep. 2015, 5, 18709. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Sun, W.; Hao, X.; Wei, M.; Su, X.; Zhang, Y.; Su, L.; Liu, X. EHMT2 inhibitor BIX-01294 induces apoptosis through PMAIP1-USP9X-MCL1 axis in human bladder cancer cells. Cancer Cell Int. 2015, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Artal-Martinez de Narvajas, A.; Gomez, T.S.; Zhang, J.S.; Mann, A.O.; Taoda, Y.; Gorman, J.A.; Herreros-Villanueva, M.; Gress, T.M.; Ellenrieder, V.; Bujanda, L.; et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol. Cell. Biol. 2013, 33, 3983–3993. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, Q.; Paulk, J.; Kubicek, S.; Kemp, M.M.; Adams, D.J.; Shamji, A.F.; Wagner, B.K.; Schreiber, S.L. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem. Biol. 2012, 7, 1152–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Tang, A.J.; Castoreno, A.B.; Kuo, S.Y.; Wang, Q.; Kuballa, P.; Xavier, R.; Shamji, A.F.; Schreiber, S.L.; Wagner, B.K. Gossypol and an HMT G9a inhibitor act in synergy to induce cell death in pancreatic cancer cells. Cell Death Dis. 2013, 4, e690. [Google Scholar] [CrossRef] [PubMed]
- Pappano, W.N.; Guo, J.; He, Y.; Ferguson, D.; Jagadeeswaran, S.; Osterling, D.J.; Gao, W.; Spence, J.K.; Pliushchev, M.; Sweis, R.F.; et al. The histone methyltransferase inhibitor A-366 uncovers a role for G9a/GLP in the epigenetics of leukemia. PLoS ONE 2015, 10, e0131716. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.R.; Hsu, M.C.; Luo, C.W.; Chen, L.T.; Shan, Y.S.; Hung, W.C. The histone methyltransferase G9a as a therapeutic target to override gemcitabine resistance in pancreatic cancer. Oncotarget 2016, 7, 61136–61151. [Google Scholar] [CrossRef] [PubMed]
- Van Vlerken, L.E.; Kiefer, C.M.; Morehouse, C.; Li, Y.; Groves, C.; Wilson, S.D.; Yao, Y.; Hollingsworth, R.E.; Hurt, E.M. EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl. Med. 2013, 2, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Miele, E.; Valente, S.; Alfano, V.; Silvano, M.; Mellini, P.; Borovika, D.; Marrocco, B.; Po, A.; Besharat, Z.M.; Catanzaro, G.; et al. The histone methyltransferase EZH2 as a druggable target in shh medulloblastoma cancer stem cells. Oncotarget 2017, 8, 68557–68570. [Google Scholar] [CrossRef] [PubMed]
- Vaswani, R.G.; Gehling, V.S.; Dakin, L.A.; Cook, A.S.; Nasveschuk, C.G.; Duplessis, M.; Iyer, P.; Balasubramanian, S.; Zhao, F.; Good, A.C.; et al. Identification of (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1h-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-cell lymphomas. J. Med. Chem. 2016, 59, 9928–9941. [Google Scholar] [PubMed]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Zhu, F.; Lin, W.R.; Ying, R.B.; Yang, Y.P.; Zeng, L.H. The novel EZH2 inhibitor, GSK126, suppresses cell migration and angiogenesis via down-regulating VEGF-A. Cancer Chemother. Pharmacol. 2016, 77, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of eed. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Curry, E.; Green, I.; Chapman-Rothe, N.; Shamsaei, E.; Kandil, S.; Cherblanc, F.L.; Payne, L.; Bell, E.; Ganesh, T.; Srimongkolpithak, N.; et al. Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin. Epigenet. 2015, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Mathison, A.; Salmonson, A.; Missfeldt, M.; Bintz, J.; Williams, M.; Kossak, S.; Nair, A.; de Assuncao, T.M.; Christensen, T.; Buttar, N.; et al. Combined aurka and H3K9 methyltransferase targeting inhibits cell growth by inducing mitotic catastrophe. Mol. Cancer Res. MCR 2017, 15, 984–997. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Jackson, S.P. G9a inhibition potentiates the anti-tumour activity of DNA double-strand break inducing agents by impairing DNA repair independent of p53 status. Cancer Lett. 2016, 380, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.W.; Mody, H.; Marrache, S.; Bhutia, Y.D.; Davis, F.; Cho, J.H.; Zastre, J.; Dhar, S.; Chu, C.K.; Govindarajan, R. Pharmacological reversal of histone methylation presensitizes pancreatic cancer cells to nucleoside drugs: In vitro optimization and novel nanoparticle delivery studies. PLoS ONE 2013, 8, e71196. [Google Scholar] [CrossRef] [PubMed]
- Kempinska, K.; Malik, B.; Borkin, D.; Klossowski, S.; Shukla, S.; Miao, H.; Wang, J.; Cierpicki, T.; Grembecka, J. Pharmacologic inhibition of the Menin-MLL interaction leads to transcriptional repression of PEG10 and blocks hepatocellular carcinoma. Mol. Cancer Ther. 2017, 17, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Kura Oncology. Kura Oncology Receives FDA Clearance to Proceed with Clinical Trial for ERK Inhibitor KO-947 and Nominates KO-539 as Development Candidate for Menin-MLL Inhibitor Program. Available online: https://globenewswire.com/news-release/2017/01/04/903160/0/en/Kura-Oncology-Receives-FDA-Clearance-to-Proceed-with-Clinical-Trial-for-ERK-Inhibitor-KO-947-and-Nominates-KO-539-as-Development-Candidate-for-Menin-MLL-Inhibitor-Program.html (accessed on 9 March 2018).
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Gaddis, M.; Gerrard, D.; Frietze, S.; Farnham, P.J. Altering cancer transcriptomes using epigenomic inhibitors. Epigenet. Chromatin 2015, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Wang, W.H.; Wu, W.Y.; Hsu, C.C.; Wei, L.R.; Wang, S.F.; Hsu, Y.W.; Liaw, C.C.; Tsai, W.C. Novel histone deacetylase inhibitor AR-42 exhibits antitumor activity in pancreatic cancer cells by affecting multiple biochemical pathways. PLoS ONE 2017, 12, e0183368. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, P.; Seidler, B.; Schuler, S.; Schnieke, A.; Gottlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Bian, B.; Bigonnet, M.; Gayet, O.; Loncle, C.; Maignan, A.; Gilabert, M.; Moutardier, V.; Garcia, S.; Turrini, O.; Delpero, J.R.; et al. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the BET bromodomain inhibitor JQ1: Implications for individualized medicine efforts. EMBO Mol. Med. 2017, 9, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, A.; Adams, C.E.; Huang, Y.; Hamarneh, S.R.; Liu, W.; Von Alt, K.N.; Mino-Kenudson, M.; Hodin, R.A.; Lillemoe, K.D.; Fernandez-Del Castillo, C.; et al. Selective and reversible suppression of intestinal stem cell differentiation by pharmacological inhibition of BET bromodomains. Sci. Rep. 2016, 6, 20390. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Li, Y.; Muqbil, I.; Aboukameel, A.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Shacham, S.; Kauffman, M.G.; Philip, P.A.; et al. Exportin 1 (XPO1) inhibition leads to restoration of tumor suppressor miR-145 and consequent suppression of pancreatic cancer cell proliferation and migration. Oncotarget 2017, 8, 82144–82155. [Google Scholar] [CrossRef] [PubMed]
- Sweis, R.F.; Pliushchev, M.; Brown, P.J.; Guo, J.; Li, F.; Maag, D.; Petros, A.M.; Soni, N.B.; Tse, C.; Vedadi, M.; et al. Discovery and development of potent and selective inhibitors of histone methyltransferase G9a. ACS Med. Chem. Lett. 2014, 5, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Mao, H.; Cao, Z.; Wang, Y.A.; Peng, X.; Wang, X.; Sajja, H.K.; Wang, L.; Duan, H.; Ni, C.; et al. Molecular imaging of pancreatic cancer in an animal model using targeted multifunctional nanoparticles. Gastroenterology 2009, 136, 1514.e2–1525.e2. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, B.; Robbins, D.H.; Lewin, D.N.; Mikhitarian, K.; Graham, A.; Rumpp, L.; Glenn, T.; Gillanders, W.E.; Cole, D.J.; et al. Accurate discrimination of pancreatic ductal adenocarcinoma and chronic pancreatitis using multimarker expression data and samples obtained by minimally invasive fine needle aspiration. Int. J. Cancer 2007, 120, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.T.; Roy, I.; Swihart, M.T.; Prasad, P.N. Multifunctional nanoparticles as biocompatible targeted probes for human cancer diagnosis and therapy. J. Mater. Chem. 2009, 19, 4655–4672. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Ke, X.; He, Z.; Yang, D.; Gong, H.; Zhang, Y.; Jing, X.; Yao, J.; Chen, J. A MSLN-targeted multifunctional nanoimmunoliposome for MRI and targeting therapy in pancreatic cancer. Int. J. Nanomed. 2012, 7, 5053–5065. [Google Scholar]
- Javle, M.M.; Varadhachary, G.R.; Fogelman, D.R.; Shroff, R.T.; Overman, M.J.; Ukegbu, L.; Bekele, B.N.; Kar, S.P.; Wolff, R.A.; Abbruzzese, J.L. Randomized phase II study of gemcitabine (G) plus anti-IGF-1R antibody MK-0646, G plus erlotinib (E) plus MK-0646 and G plus E for advanced pancreatic cancer. J. Clin. Oncol. 2011, 29, 4026. [Google Scholar] [CrossRef]


{kind=link}
| Epigenetic Pathway | Enzymatic Target | Drug Name | Trial/Clinical Setting |
|---|---|---|---|
| DNA methylation | DNMT1/2 | 5-azacitidine | FDA approved (myelodysplastic syndromes) |
| DNMT1 | RG-108 derivatives | ||
| H3K4me | Menin (MLL binding) | MI-503 | |
| Menin (MLL binding) | KO-539 | ||
| KDM5 | CPI-445 | ||
| LSD (KDM1A) | GSK2879552 | Trial NTC02929498; recruiting | |
| Tranylcypromine | FDA approved (depression) | ||
| H3K9me | G9a | BRD-4770 | |
| A-366 | |||
| BIX-01294 | |||
| UNC0638 | |||
| SUV39H1 | Chaetocin | ||
| H3K27me | EZH2 | CPI-1205 | Trial NCT02395601; Phase I; accruing |
| UNC1999 | |||
| GSK126 | |||
| Tazemetostat | Trial NCT03009344; NCT02860286; both active, not recruiting | ||
| demethylating agent | 3-deazaneplanocin A | ||
| H3K27Ac | HDAC | AR-42 | Tirals NCT02795819; NCT01798901; NCT01129193; all accruing |
| CG200745 | Trials NTC02737228; NCT02737462; both recruiting | ||
| CBP | ICG-001 | ||
| BET family | JQ1 | ||
| I-BET 762 | |||
| CPI-203 | |||
| miRNA-122 | Hepatitis C Virus | Miravirsen | Trial NCT02508090; Phase II; complete |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paradise, B.D.; Barham, W.; Fernandez-Zapico, M.E. Targeting Epigenetic Aberrations in Pancreatic Cancer, a New Path to Improve Patient Outcomes? Cancers 2018, 10, 128. https://doi.org/10.3390/cancers10050128
Paradise BD, Barham W, Fernandez-Zapico ME. Targeting Epigenetic Aberrations in Pancreatic Cancer, a New Path to Improve Patient Outcomes? Cancers. 2018; 10(5):128. https://doi.org/10.3390/cancers10050128
Chicago/Turabian StyleParadise, Brooke D., Whitney Barham, and Martín E. Fernandez-Zapico. 2018. "Targeting Epigenetic Aberrations in Pancreatic Cancer, a New Path to Improve Patient Outcomes?" Cancers 10, no. 5: 128. https://doi.org/10.3390/cancers10050128
APA StyleParadise, B. D., Barham, W., & Fernandez-Zapico, M. E. (2018). Targeting Epigenetic Aberrations in Pancreatic Cancer, a New Path to Improve Patient Outcomes? Cancers, 10(5), 128. https://doi.org/10.3390/cancers10050128