A Phase II Study of Pelareorep (REOLYSIN®) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma



Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
2.2. Efficacy
2.3. Toxicity
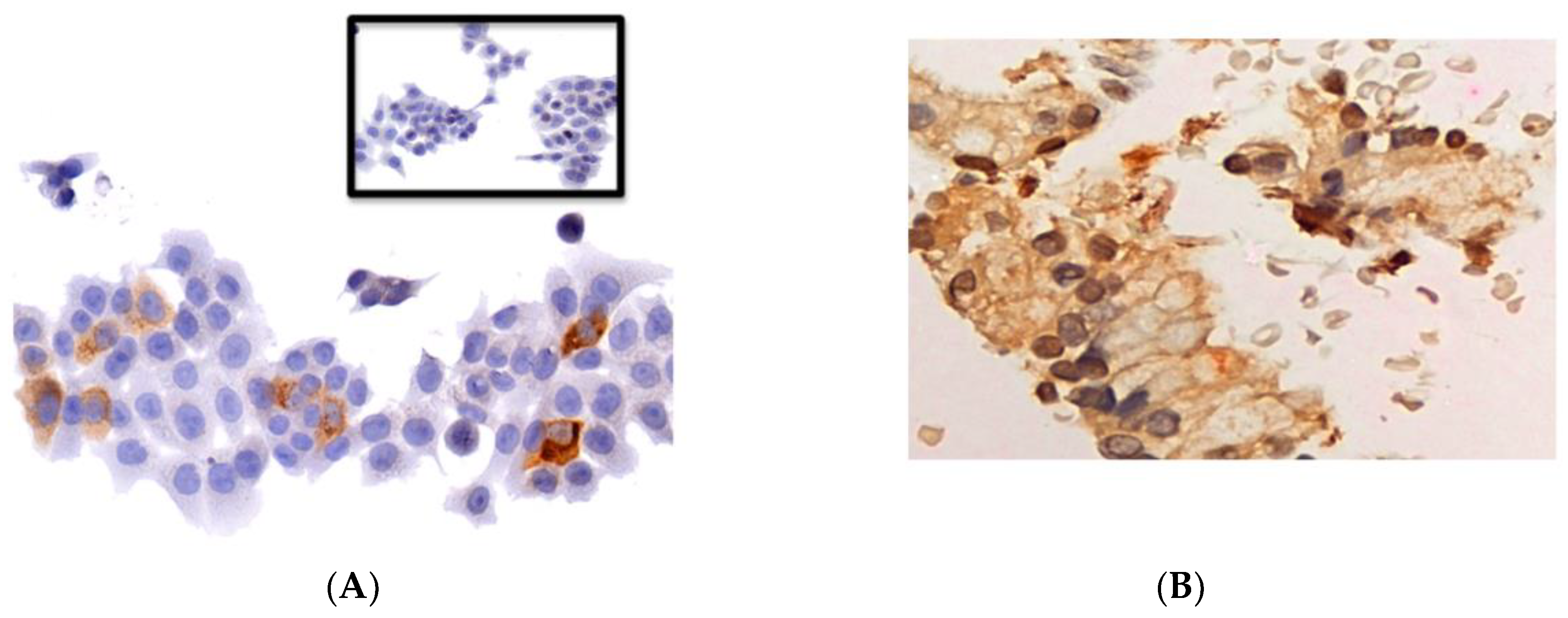
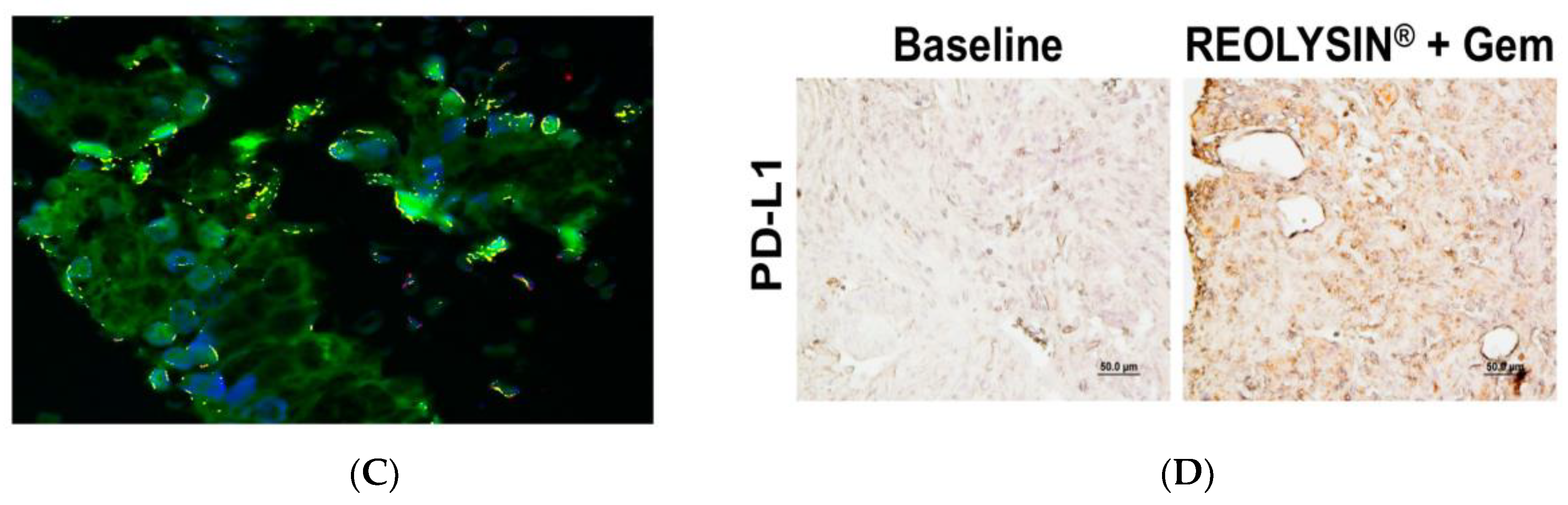
2.4. Pharmacodynamic Analysis
3. Discussion
4. Materials and Methods
4.1. Ethical Considerations
4.2. Study Design and Treatment
4.3. Study Endpoints
4.4. Study Assessments
4.5. Safety Evaluations
4.6. Pharmacodynamic Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. Folfirinox versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Rubinson, D.A.; Wolpin, B.M. Therapeutic approaches for metastatic pancreatic adenocarcinoma. Hematol. Oncol. Clin. N. Am. 2015, 29, 761–776. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): Pancreatic Adenocarcinoma, Version 2 ed.; National Comprehensive Cancer Network: Jenkintown, PA, USA, 2015. [Google Scholar]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. Kras: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Haas, M.; Boeck, S. Systemic treatment of advanced pancreatic cancer. Cancer Treat. Rev. 2012, 38, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Mita, M.M. Activated ras signaling pathways and reovirus oncolysis: An update on the mechanism of preferential reovirus replication in cancer cells. Front. Oncol. 2014, 4, 167. [Google Scholar] [CrossRef] [PubMed]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.L.; Hirasawa, K.; Yang, A.D.; Shields, M.A.; Lee, P.W. Reovirus oncolysis: The Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 11099–11104. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Pan, L.Z.; Hill, R.; Marcato, P.; Shmulevitz, M.; Vassilev, L.T.; Lee, P.W. Stabilisation of p53 enhances reovirus-induced apoptosis and virus spread through p53-dependent NF-kappaB activation. Br. J. Cancer 2011, 105, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Sei, S.; Yang, Q.E.; Mussio, J.K.; Coffey, M.C.; Parchment, J.E.; Shoemaker, R.H.; Tomaszewski, J.E. Synergistic antitumor activity of oncolytic reovirus and chemotherapeutic agents against non-small cell lung cancer. Eur. J. Cancer Suppl. 2006, 4, 103. [Google Scholar] [CrossRef]
- Wadler, S.; Yu, B.; Lane, M.; Klampfer, L.; Sasazuki, T.; Shirasawa, S.; Coffey, M. The oncolytic reovirus, reolysin, augments the anticancer effects of cytotoxic agents in vitro against the ras-mutated human colon cancer cell line hct116. Eur. J. Cancer Suppl. 2004, 2, 135. [Google Scholar] [CrossRef]
- Errington, F.; Steele, L.; Prestwich, R.; Harrington, K.J.; Pandha, H.S.; Vidal, L.; de Bono, J.; Selby, P.; Coffey, M.; Vile, R.; et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J. Immunol. 2008, 180, 6018–6026. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Steele, L.P.; Ilett, E.J.; Morgan, R.S.; Harrington, K.J.; Pandha, H.S.; Selby, P.J.; Vile, R.G.; Melcher, A.A. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J. Immunol. 2009, 183, 4312–4321. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Ilett, E.J.; Errington, F.; Diaz, R.M.; Steele, L.P.; Kottke, T.; Thompson, J.; Galivo, F.; Harrington, K.J.; Pandha, H.S.; et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin. Cancer Res. 2009, 15, 4374–4381. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.A.; Lee, P.W. Oncolytic virus-mediated reversal of impaired tumor antigen presentation. Front. Oncol. 2014, 4, 77. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.A.; Marcato, P.; Pan, D.; Lee, P.W. Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Mol. Cancer Ther. 2010, 9, 2924–2933. [Google Scholar] [CrossRef] [PubMed]
- Adair, R.A.; Scott, K.J.; Fraser, S.; Errington-Mais, F.; Pandha, H.; Coffey, M.; Selby, P.; Cook, G.P.; Vile, R.; Harrington, K.J.; et al. Cytotoxic and immune-mediated killing of human colorectal cancer by reovirus-loaded blood and liver mononuclear cells. Int. J. Cancer 2013, 132, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Noonan, A.M.; Farren, M.R.; Geyer, S.M.; Huang, Y.; Tahiri, S.; Ahn, D.; Mikhail, S.; Ciombor, K.K.; Pant, S.; Aparo, S.; et al. Randomized phase 2 trial of the oncolytic virus pelareorep (reolysin) in upfront treatment of metastatic pancreatic adenocarcinoma. Mol. Ther. 2016, 24, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Rajani, K.; Parrish, C.; Kottke, T.; Thompson, J.; Zaidi, S.; Ilett, L.; Shim, K.G.; Diaz, R.M.; Pandha, H.; Harrington, K.; et al. Combination therapy with reovirus and Anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol. Ther. 2016, 24, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Etoh, T.; Himeno, Y.; Matsumoto, T.; Aramaki, M.; Kawano, K.; Nishizono, A.; Kitano, S. Oncolytic viral therapy for human pancreatic cancer cells by reovirus. Clin. Cancer Res. 2003, 9, 1218–1223. [Google Scholar] [PubMed]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Kelly, K.R.; Coffey, M.; Freeman, J.W.; Nawrocki, S.T. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis. 2013, 4, e728. [Google Scholar] [CrossRef] [PubMed]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W. Reovirus therapy of tumors with activated Ras pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Lolkema, M.P.; Arkenau, H.T.; Harrington, K.; Roxburgh, P.; Morrison, R.; Roulstone, V.; Twigger, K.; Coffey, M.; Mettinger, K.; Gill, G.; et al. A phase I study of the combination of intravenous reovirus type 3 dearing and gemcitabine in patients with advanced cancer. Clin. Cancer Res. 2011, 17, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Vidal, L.; Pandha, H.S.; Yap, T.A.; White, C.L.; Twigger, K.; Vile, R.G.; Melcher, A.; Coffey, M.; Harrington, K.J.; DeBono, J.S. A phase I study of intravenous oncolytic reovirus type 3 dearing in patients with advanced cancer. Clin. Cancer Res. 2008, 14, 7127–7137. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ramanathan, R.K.; Borad, M.J.; Laheru, D.A.; Smith, L.S.; Wood, T.E.; Korn, R.L.; Desai, N.; Trieu, V.; Iglesias, J.L.; et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: A phase I/II trial. J. Clin. Oncol. 2011, 29, 4548–4554. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic cancer: Long-term survival from a phase iii trial. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Villalona-Calero, M.A.; Lam, E.; Otterson, G.A.; Zhao, W.; Timmons, M.; Subramaniam, D.; Hade, E.M.; Gill, G.M.; Coffey, M.; Selvaggi, G.; et al. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with KRAS-activated tumors. Cancer 2016, 122, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Pirker, R.; Pereira, J.R.; Szczesna, A.; von Pawel, J.; Krzakowski, M.; Ramlau, R.; Vynnychenko, I.; Park, K.; Yu, C.T.; Ganul, V.; et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (flex): An open-label randomised phase iii trial. Lancet 2009, 373, 1525–1531. [Google Scholar] [CrossRef]
- Bernstein, V.; Ellard, S.L.; Dent, S.F.; Tu, D.; Mates, M.; Dhesy-Thind, S.K.; Panasci, L.; Gelmon, K.A.; Salim, M.; Song, X.; et al. A randomized phase II study of weekly paclitaxel with or without pelareorep in patients with metastatic breast cancer: Final analysis of Canadian cancer trials group ind.213. Breast Cancer Res. Treat. 2018, 167, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Patel, S.; Nuovo, G.; Gill, G.; Selvaggi, G.; Coffey, M.; Nawrocki, S.T. The combination of intravenous Reolysin and gemcitabine induces reovirus replication and endoplasmic reticular stress in a patient with KRAS-activated pancreatic cancer. BMC Cancer 2015, 15, 513. [Google Scholar] [CrossRef] [PubMed]
- Saqib, R.; Ashraf, N.; Chavez, J.C.; Malafa, M.P.; Coppola, D.; Springett, G.M.; Helm, J.; Kim, R.D. Expression of programmed death ligand 1 (PD-L1) in malignant and nonmalignant pancreatic tissue. J. Clin. Oncol. 2013, 31, 215. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of Anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Annels, N.E.; Simpson, G.R.; Arif, M.; Denyer, M.; Harrington, K.; Coffey, M.; Vile, R.; Melcher, A.; Pandha, H. B and t lymphocyte attenuator (BTLA) and PD-L1 significantly upregulated in reovirus treated tramp-c2 tumours. In Proceedings of the 11th International Oncolytics Virus Conference, Oxford University, Oxford, UK, 9–12 April 2018. [Google Scholar]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, G.J.; Garofalo, M.; Valeri, N.; Roulstone, V.; Volinia, S.; Cohn, D.E.; Phelps, M.; Harrington, K.J.; Vile, R.; Melcher, A.; et al. Reovirus-associated reduction of microrna-let-7d is related to the increased apoptotic death of cancer cells in clinical samples. Mod. Pathol. 2012, 25, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Kepner, J.L.; Chang, M.N. Samples of exact k-stage group sequential designs for phase II and pilot studies. Control. Clin. Trials 2004, 25, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.; Mahalingam, D. Immunotherapy in pancreatic adenocarcinoma-overcoming barriers to response. J. Gastrointest. Oncol. 2018, 1, 143–159. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | Number or Percentage |
|---|---|
| Total number | 34 |
| Median Age (range), years | 66 (48–85) |
| ≥65 years of age, % | 53 |
| Sex, % | |
| Male | 53 |
| Female | 47 |
| ECOG Performance Status, % | |
| 0–1 | 94 |
| 2 | 6 |
| Ethnicity, % | |
| Caucasian | 71 |
| Black | 12 |
| Hispanic | 3 |
| Asian | 3 |
| Metastatic disease at baseline, % | 91 |
| Site of metastases, % | |
| Liver | 65 |
| Lung | 6 |
| Peritoneum | 18 |
| Number of metastatic sites, % | |
| 1 | 35 |
| 2 | 18 |
| 3 | 3 |
| >3 | 35 |
| Median number of cycles (schedule every 3 weeks) | 4 |
| Previous chemotherapy/radiotherapy, % | 5 |
| Therapy after disease progression on pelareorep, % | 53 |
| Toxicity * | Total % | Grade 3% | Grade 4% |
|---|---|---|---|
| Hematologic | |||
| Anemia | 35 | 24 | 3 |
| Neutropenia | 32 | 15 | 12 |
| Thrombocytopenia | 15 | 6 | 0 |
| Non-Hematologic | |||
| Diarrhoea | 24 | 0 | 0 |
| Nausea | 29 | 0 | 0 |
| Vomiting | 24 | 0 | 0 |
| Fatigue | 71 | 9 | 0 |
| Chills/Flu-like symptoms | 51 | 0 | 0 |
| Edema | 33 | 0 | 0 |
| Fever | 56 | 0 | 0 |
| AST increased | 12 | 6 | 0 |
| Anorexia/Weight loss | 33 | 0 | 0 |
| Dyspnea | 50 | 6 | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahalingam, D.; Goel, S.; Aparo, S.; Patel Arora, S.; Noronha, N.; Tran, H.; Chakrabarty, R.; Selvaggi, G.; Gutierrez, A.; Coffey, M.; et al. A Phase II Study of Pelareorep (REOLYSIN®) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma. Cancers 2018, 10, 160. https://doi.org/10.3390/cancers10060160
Mahalingam D, Goel S, Aparo S, Patel Arora S, Noronha N, Tran H, Chakrabarty R, Selvaggi G, Gutierrez A, Coffey M, et al. A Phase II Study of Pelareorep (REOLYSIN®) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma. Cancers. 2018; 10(6):160. https://doi.org/10.3390/cancers10060160
Chicago/Turabian StyleMahalingam, Devalingam, Sanjay Goel, Santiago Aparo, Sukeshi Patel Arora, Nicole Noronha, Hue Tran, Romit Chakrabarty, Giovanni Selvaggi, Andres Gutierrez, Matthew Coffey, and et al. 2018. "A Phase II Study of Pelareorep (REOLYSIN®) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma" Cancers 10, no. 6: 160. https://doi.org/10.3390/cancers10060160
APA StyleMahalingam, D., Goel, S., Aparo, S., Patel Arora, S., Noronha, N., Tran, H., Chakrabarty, R., Selvaggi, G., Gutierrez, A., Coffey, M., Nawrocki, S. T., Nuovo, G., & Mita, M. M. (2018). A Phase II Study of Pelareorep (REOLYSIN®) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma. Cancers, 10(6), 160. https://doi.org/10.3390/cancers10060160