Deregulation of Negative Controls on TGF-β1 Signaling in Tumor Progression


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to TGF-β1 Signaling
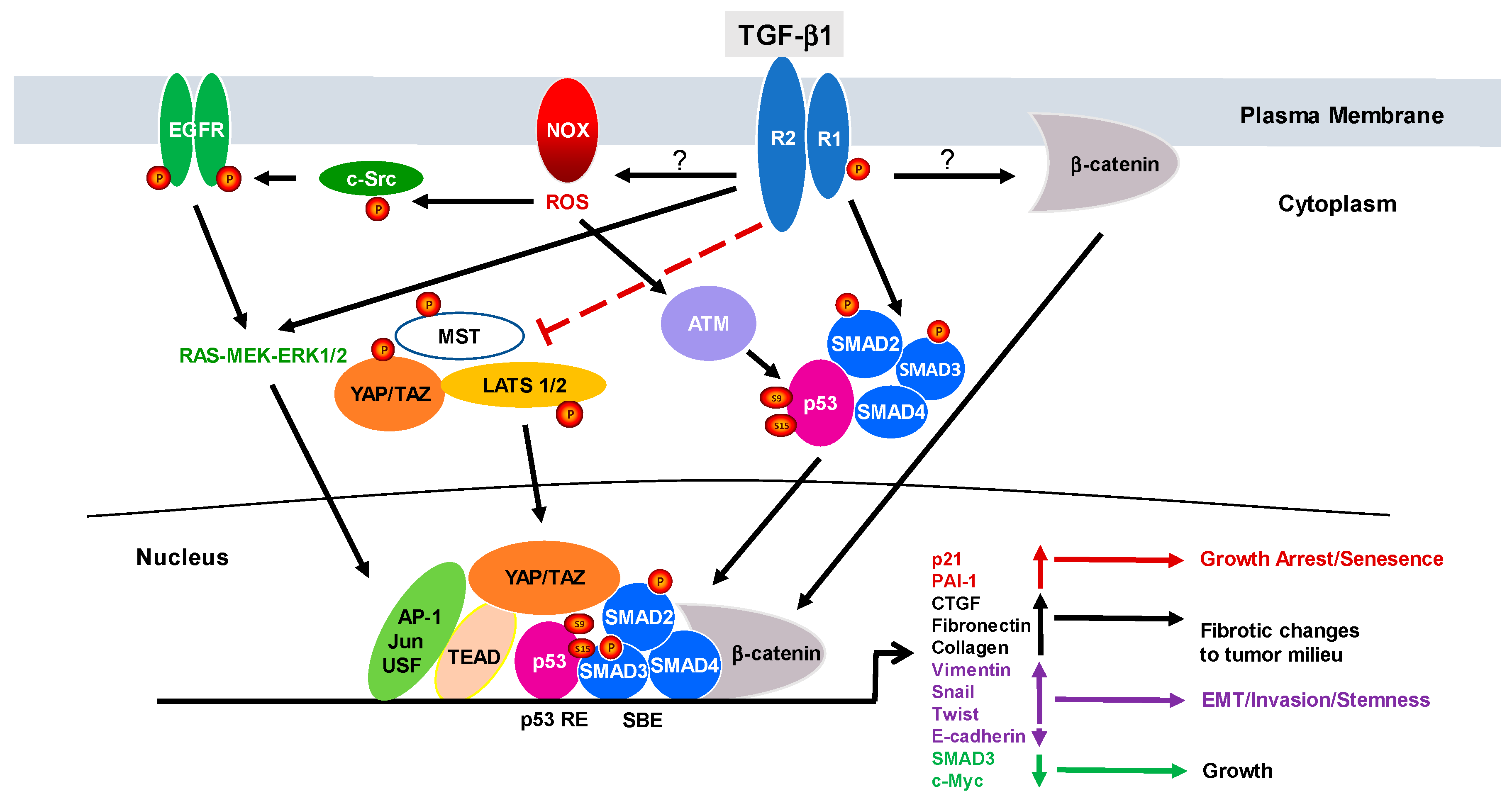
2. The Duality of TGF-β1 Signaling: From Cancer Suppressor to Tumor Promotor
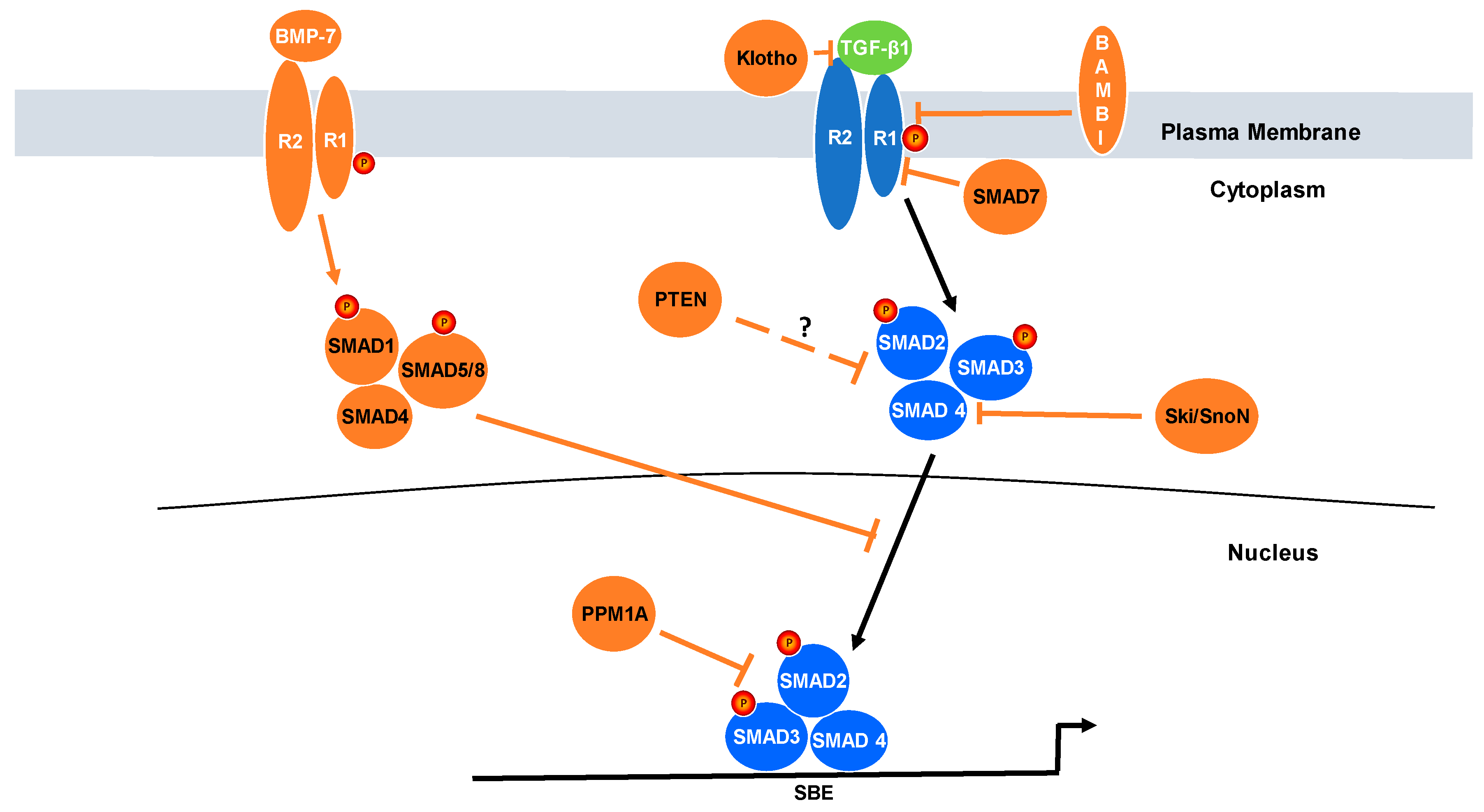
3. Relevance of Negative Regulators of TGF-β Signaling to Cancer Progression
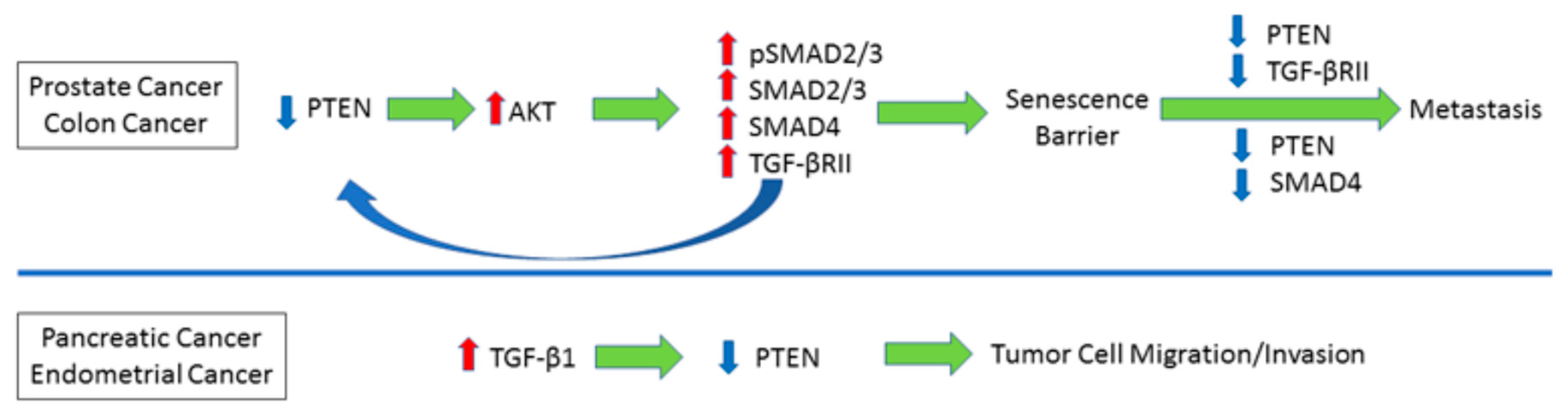
4. Reciprocal Relationship between PTEN and TGF-β Signaling
5. Protein Phosphatase Magnesium-Dependent 1A (PPM1A)
6. SMAD7
7. Klotho
8. BMP7
9. Ski/SnoN
10. Bone Morphogenetic Protein and Activin Membrane-Bound Inhibitor (BAMBI) and Rac1b
11. Pathological Significance, Perspectives and Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| AP-1 | Activator Protein 1 |
| ATM | Ataxia-telangiectasia mutated |
| BAMBI | Bone morphogenetic protein and activin membrane-bound inhibitor |
| BMP | Bone morphogenetic protein |
| CBP | CREB-binding protein |
| DNMT | DNA methyltransferases |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-mesenchymal transition |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| HCC | Hepatocellular carcinoma |
| HCV | Human hepatitis C virus |
| HECT | E6-associated protein carboxyl terminus |
| ID | Inhibitor of DNA binding |
| IGF | Insulin-like growth factor |
| MH1 | MAD homology 1 |
| MMP | Matrix metalloprotease |
| OTUD1 | OTU domain-containing protein 1 |
| P/CAF | P300/CBP-associated factor |
| PDAC | Pancreatic ductal adenocarcinoma |
| PIP2 | Phosphatidylinositol-4,5-triphosphate |
| PIP3 | Phosphatidylinositol-3,4,5-triphosphate |
| PI3K | Phosphoinositide 3-kinase |
| PPM1A | Protein phosphatase magnesium/ manganese-dependent 1A |
| PTEN | Phosphatase and tensin homologue |
| R-SMAD | Receptor-regulated SMAD |
| ROS | Reactive oxygen species |
| Ski | Sloan-Kettering Institute proto-oncogene |
| SMAD | Mothers against decapentaplegic homolog |
| Smurf | SMAD ubiquitination regulatory factors |
| SnoN | Ski related novel gene |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| TEAD | TEA domain family |
| TGF-β | Transforming growth factor β |
| TGF-βR | Transforming growth factor receptor |
| USF | Upstream stimulatory factor |
| USP26 | Ubiquitin-specific protease 26 |
| UUO | Unilateral ureteral obstruction |
| YAP | Yes-associated protein |
References
- Massague, J. TGF-β signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; Ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Abdollah, S.; Macias-Silva, M.; Tsukazaki, T.; Hayashi, H.; Attisano, L.; Wrana, J.L. TβRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 1997, 272, 27678–27685. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, X.; We, R.; Derynck, R. Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature 1996, 383, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.H.; Miyazono, K.; et al. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, M.; Kanduri, M.; Gronroos, E.; Heldin, C.H.; Ericsson, J. The DNA binding activities of Smad2 and Smad3 are regulated by coactivator-mediated acetylation. J. Biol. Chem. 2006, 281, 39870–39880. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Overstreet, J.M.; Higgins, P.J. TGF-β signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cell. Signal. 2013, 25, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Higgins, P.J. Integration of non-SMAD and SMAD signaling in TGF-β1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb. Haemost. 2008, 100, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Higgins, S.P.; Higgins, C.E.; Higgins, P.J. TGF-β1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscle cells requires pp60c-src/EGFRY845 and Rho/ROCK signaling. J. Mol. Cell. Cardiol. 2008, 44, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Dobberfuhl, A.D.; Cooley, C.; Overstreet, J.M.; Patel, S.; Goldschmeding, R.; Meldrum, K.K.; Higgins, P.J. Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell. Signal. 2013, 25, 2198–2209. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, J.M.; Samarakoon, R.; Cardona-Grau, D.; Goldschmeding, R.; Higgins, P.J. Tumor suppressor ataxia telangiectasia mutated functions downstream of TGF-β1 in orchestrating profibrotic responses. FASEB J. 2015, 29, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, J.M.; Samarakoon, R.; Meldrum, K.K.; Higgins, P.J. Redox control of p53 in the transcriptional regulation of TGF-β1 target genes through SMAD cooperativity. Cell. Signal. 2014, 26, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Chitnis, S.S.; Higgins, S.P.; Higgins, C.E.; Krepinsky, J.C.; Higgins, P.J. Redox-induced Src kinase and caveolin-1 signaling in TGF-β1-initiated SMAD2/3 activation and PAI-1 expression. PLoS ONE 2011, 6, e22896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, X.H.; Derynck, R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature 1998, 394, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Anorga, S.; Overstreet, J.M.; Falke, L.L.; Tang, J.; Goldschmeding, R.G.; Higgins, P.J.; Samarakoon, R. Deregulation of Hippo-TAZ pathway during renal injury confers a fibrotic maladaptive phenotype. FASEB J. 2018, 32, 2644–2657. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell. Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Guan, K.L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Toyoda, T.; Nakanishi, H.; Yatabe, Y.; Sato, A.; Matsudaira, Y.; Ito, H.; Murakami, H.; Kondo, Y.; Kondo, E.; et al. TGF-β synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth. J. Exp. Med. 2012, 209, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, J.-J. The dual role of TGFβ in human cancer: From tumor suppression to cancer metastasis. ISRN Mol. Biol. 2012, 2012, 381428. [Google Scholar] [CrossRef] [PubMed]
- Bartholin, L.; Vincent, D.F.; Valcourt, U. TGF-β as a tumor suppressor: In vitro mechanistic aspects of growth inhibition. In TGFβ in Human Disease; Moustakas, A., Miyazawa, K., Eds.; Springer: Tokyo, Japan, 2013; pp. 113–138. [Google Scholar]
- Valcourt, U.; Vincent, D.F.; Bartholin, L. TGF-β as tumor suppressor: Lessons from mouse models. In TGFβ in Human Disease; Moustakas, A., Miyazawa, K., Eds.; Springer: Tokyo, Japan, 2013; pp. 139–168. [Google Scholar]
- Wendt, M.K.; Schiemann, W.P. The multifunctional roles of TGF-β in navigating the metastatic cascade. In TGFβ in Human Disease; Moustakas, A., Miyazawa, K., Eds.; Springer: Tokyo, Japan, 2013; pp. 169–187. [Google Scholar]
- Siegal, P.M.; Massague, J. Growth control by TGF-β: Mechanisms controlling cell cycle progression and apoptosis. In The TGF-β Family; Derynck, R., Miyazone, K., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2008; pp. 333–362. [Google Scholar]
- Schilling, S.H.; Hjelmeland, A.B.; Rich, J.N.; Wang, X.F. TGF-β: A multipotential cytokine. In The TGF-β Family; Derynck, R., Miyazone, K., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2008; pp. 45–77. [Google Scholar]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Hachim, I.Y.; Hachim, M.Y.; Lopez-Ozuna, V.M.; Lebrun, J.J. A dual prognostic role for the TGFβ receptors in human breast cancer. Hum. Pathol. 2016, 57, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Caja, F.; Vannucci, L. TGFβ: A player on multiple fronts in the tumor microenvironment. J. Immunotox. 2015, 12, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumor microenvironment. Nat. Rev. Cancer 2013, 13, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; ten Dijke, P. Negative regulation of TGF-β receptor/Smad signal transduction. Curr. Opin. Cell Biol. 2007, 19, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell. Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofano, A.; Pandolfi, P.P. The multiple roles of PTEN in tumor suppression. Cell 2000, 100, 387–390. [Google Scholar] [CrossRef]
- Shariat, S.F.; Shalev, M.; Menesses-Diaz, A.; Kim, I.Y.; Kattan, M.W.; Wheeler, T.M.; Slawin, K.M. Preoperative plasma levels of transforming growth factor β1 (TGF-β1) strongly predict progression in patients undergoing radical prostatectomy. J. Clin. Oncol. 2001, 19, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wu, C.J.; Chu, G.C.; Xiao, Y.; Ho, D.; Zhang, J.; Perry, S.R.; Labrot, E.S.; Wu, X.; Lis, R.; et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011, 470, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Niki, M.; Dotan, Z.A.; Koutcher, J.A.; Di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; Van Dyke, T.; et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003, 1, e59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerke, G.A.; Yang, C.S.; Frierson, H.F.; Paschal, B.M.; Wotton, D. Activation of Akt signaling in prostate induces a TGFβ-mediated restraint on cancer progression and metastasis. Oncogene 2014, 33, 3660–3667. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Helo, S.; Dobberfuhl, A.D.; Khakoo, N.S.; Falke, L.; Overstreet, J.M.; Goldschmeding, R.; Higgins, P.J. Loss of tumour suppressor PTEN expression in renal injury initiates SMAD3- and p53-dependent fibrotic responses. J. Pathol. 2015, 236, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Trobridge, P.; Wang, Y.; Kanngurn, S.; Morris, S.M.; Knoblaugh, S.; Grady, W.M. Inactivation of TGF-β signaling and loss of PTEN cooperate to induce colon cancer in vivo. Oncogene 2014, 33, 1538–1547. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Yamanaka, Y.; Buchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor β isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar] [CrossRef]
- Ebert, M.P.; Fei, G.; Schandl, L.; Mawrin, C.; Dietzmann, K.; Herrera, P.; Friess, H.; Gress, T.M.; Malfertheiner, P. Reduced PTEN expression in the pancreas overexpressing transforming growth factor-β1. Br. J. Cancer 2002, 86, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.Y.; Dong, H.; Quach, K.T.; Van Nguyen, P.N.; Chen, K.; Carethers, J.M. TGF-β mediates PTEN suppression and cell motility through calcium-dependent PKC-α activation in pancreatic cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G899–G905. [Google Scholar] [CrossRef] [PubMed]
- Yabushita, H.; Narumiya, H.; Hiratake, K.; Yamada, H.; Shimazu, M.; Sawaguchi, K.; Noguchi, M.; Nakanishi, M. The association of transforming growth factor-β1 with myometrial invasion of endometrial carcinomas through effects on matrix metalloproteinase. J. Obstet. Gynaecol. Res. 2000, 26, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Cheng, J.C.; Klausen, C.; Zhao, J.; Leung, P.C. TGF-β1 stimulates migration of type II endometrial cancer cells by down-regulating PTEN via activation of SMAD and ERK1/2 signaling pathways. Oncotarget 2016, 7, 61262–61272. [Google Scholar] [PubMed]
- Mann, D.J.; Campbell, D.G.; McGowan, C.H.; Cohen, P.T. Mammalian protein serine/threonine phosphatase 2C: cDNA cloning and comparative analysis of amino acid sequences. Biochim. Biophys. Acta 1992, 1130, 100–104. [Google Scholar] [CrossRef]
- Lammers, T.; Lavi, S. Role of type 2C protein phosphatases in growth regulation and in cellular stress signaling. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Duan, X.; Liang, Y.Y.; Su, Y.; Wrighton, K.H.; Long, J.; Hu, M.; Davis, C.M.; Wang, J.; Brunicardi, F.C.; et al. PPM1A functions as a Smad phosphatase to terminate TGFβ signaling. Cell 2006, 125, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Fan, J.; Ouyang, Q.; Zhang, X.; Zhang, X.; Yu, J.; Xu, Z.; Li, Q.; Yao, X.; Liu, X.; et al. Loss of PPM1A expression enhances invasion and the epithelial-to- mesenchymal transition in bladder cancer by activating the TGF-β/Smad signaling pathway. Oncotarget 2014, 5, 5700–5711. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, Y.; Gao, Y.; Hu, W.; Qu, Y.; Lou, N.; Zhu, Y.; Zhang, X.; Yang, H. Hepatitis C virus NS3 protein enhances hepatocellular carcinoma cell invasion by promoting PPM1A ubiquitination and degradation. J. Exp. Clin. Cancer Res. 2017, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; An, H.; Jin, R.; Zou, M.; Guo, Y.; Su, P.F.; Liu, D.; Shyr, Y.; Yarbrough, W.G. PPM1A is a RelA phosphatase with tumor suppressor-like activity. Oncogene 2014, 33, 2918–2927. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Yang, M.X.; Ouyang, Q.; Fu, D.; Xu, Z.; Liu, X.; Mino-Kenudson, M.; Geng, J.; Tang, F. Phosphatase PPM1A is a novel prognostic marker in pancreatic ductal adenocarcinoma. Hum. Pathol. 2016, 55, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Rehfuss, A.; Khakoo, N.S.; Falke, L.L.; Dobberfuhl, A.D.; Helo, S.; Overstreet, J.M.; Goldschmeding, R.; Higgins, P.J. Loss of expression of protein phosphatase magnesium-dependent 1A during kidney injury promotes fibrotic maladaptive repair. FASEB J. 2016, 30, 3308–3320. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 1997, 389, 631–635. [Google Scholar] [PubMed]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF β receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Stolfi, C.; Marafini, I.; De Simone, V.; Pallone, F.; Monteleone, G. The dual role of Smad7 in the control of cancer growth and metastasis. Int. J. Mol. Sci. 2013, 14, 23774–23790. [Google Scholar] [CrossRef] [PubMed]
- Rodeck, U.; Melber, K.; Kath, R.; Menssen, H.D.; Varello, M.; Atkinson, B.; Herlyn, M. Constitutive expression of multiple growth factor genes by melanoma cells but not normal melanocytes. J. Investig. Dermatol. 1991, 97, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Delmas, V.; Moller, M.; Sextius, P.; Andre, J.; Menashi, S.; Larue, L.; Mauviel, A. Stable overexpression of Smad7 in human melanoma cells inhibits their tumorigenicity in vitro and in vivo. Oncogene 2005, 24, 7624–7629. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Mohammad, K.S.; McKenna, C.R.; Fournier, P.; Luciani, F.; Niewolna, M.; Andre, J.; Delmas, V.; Larue, L.; Guise, T.A.; et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 2007, 67, 2317–2324. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFβ signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Fan, Y.; Xie, F.; Zhou, H.; Jin, K.; Shao, L.; Shi, W.; Fang, P.; Yang, B.; van Dam, H.; et al. Breast cancer metastasis suppressor OTUD1 deubiquitinates SMAD7. Nat. Commun. 2017, 8, 2116. [Google Scholar] [CrossRef] [PubMed]
- Kit Leng Lui, S.; Iyengar, P.V.; Jaynes, P.; Isa, Z.; Pang, B.; Tan, T.Z.; Eichhorn, P.J.A. USP26 regulates TGF-β signaling by deubiquitinating and stabilizing SMAD7. EMBO Rep. 2017, 18, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Boulay, J.L.; Mild, G.; Lowy, A.; Reuter, J.; Lagrange, M.; Terracciano, L.; Laffer, U.; Herrmann, R.; Rochlitz, C. SMAD7 is a prognostic marker in patients with colorectal cancer. Int. J. Cancer 2003, 104, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Broderick, P.; Carvajal-Carmona, L.; Pittman, A.M.; Webb, E.; Howarth, K.; Rowan, A.; Lubbe, S.; Spain, S.; Sullivan, K.; Fielding, S.; et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat. Genet. 2007, 39, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Stolfi, C.; De Simone, V.; Colantoni, A.; Franze, E.; Ribichini, E.; Fantini, M.C.; Caruso, R.; Monteleone, I.; Sica, G.S.; Sileri, P.; et al. A functional role for Smad7 in sustaining colon cancer cell growth and survival. Cell Death Dis. 2014, 5, e1073. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. Smad7 induces tumorigenicity by blocking TGF-β-induced growth inhibition and apoptosis. Exp. Cell Res. 2005, 307, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Rachakonda, G.; Deane, N.G.; Datta, P.K. Smad7 induces hepatic metastasis in colorectal cancer. Br. J. Cancer 2008, 99, 957–965. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Li, A.G.; Wang, D.; Han, S.; Zheng, B.; Goumans, M.J.; Ten Dijke, P.; Wang, X.J. Overexpression of Smad7 results in severe pathological alterations in multiple epithelial tissues. EMBO J. 2002, 21, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Kuang, C.; Xiao, Y.; Liu, X.; Stringfield, T.M.; Zhang, S.; Wang, Z.; Chen, Y. In vivo disruption of TGF-β signaling by Smad7 leads to premalignant ductal lesions in the pancreas. Proc. Natl. Acad. Sci. USA 2006, 103, 1858–1863. [Google Scholar] [CrossRef] [PubMed]
- Segawa, H.; Yamanaka, S.; Ohno, Y.; Onitsuka, A.; Shiozawa, K.; Aranami, F.; Furutani, J.; Tomoe, Y.; Ito, M.; Kuwahata, M.; et al. Correlation between hyperphosphatemia and type II Na-Pi cotransporter activity in klotho mice. Am. J. Physiol. Renal. Physiol. 2007, 292, F769–F779. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, Y.; Zhou, D.; Tan, R.J.; Liu, Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/β-catenin signaling. J. Am. Soc. Nephrol. 2013, 24, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Nie, L.; He, T.; Yang, K.; Xiao, T.; Wang, S.; Huang, Y.; Zhang, J.; Wang, J.; Sharma, K.; et al. Klotho suppresses renal tubulo-interstitial fibrosis by controlling basic fibroblast growth factor-2 signalling. J. Pathol. 2014, 234, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, K.; Amin, R.; Moe, O.W.; Hu, M.C.; Erben, R.G.; Ostman Wernerson, A.; Lanske, B.; Olauson, H.; Larsson, T.E. The kidney is the principal organ mediating klotho effects. J. Am. Soc. Nephrol. 2014, 25, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho inhibits transforming growth factor-β1 (TGF-β1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M. Klotho: A master regulator of cardiovascular disease? Circulation 2012, 125, 2181–2183. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, Y. Loss of Klotho in CKD Breaks One’s Heart. J. Am. Soc. Nephrol. 2015, 26, 2305–2307. [Google Scholar] [CrossRef] [PubMed]
- Donate-Correa, J.; Martin-Nunez, E.; Mora-Fernandez, C.; Muros-de-Fuentes, M.; Perez-Delgado, N.; Navarro-Gonzalez, J.F. Klotho in cardiovascular disease: Current and future perspectives. World J. Biol. Chem. 2015, 6, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Mencke, R.; Olauson, H.; Hillebrands, J.L. Effects of Klotho on fibrosis and cancer: A renal focus on mechanisms and therapeutic strategies. Adv. Drug. Deliv. Rev. 2017, 121, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Rubinek, T.; Shulman, M.; Israeli, S.; Bose, S.; Avraham, A.; Zundelevich, A.; Evron, E.; Gal-Yam, E.N.; Kaufman, B.; Wolf, I. Epigenetic silencing of the tumor suppressor klotho in human breast cancer. Breast Cancer Res. Treat. 2012, 133, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, X.; Wang, X.; Jie, P.; Lu, H.; Zhang, S.; Lin, X.; Lam, E.K.; Cui, Y.; Yu, J. Klotho is silenced through promoter hypermethylation in gastric cancer. Am. J. Cancer. Res. 2011, 1, 111–119. [Google Scholar] [PubMed]
- Pan, J.; Zhong, J.; Gan, L.H.; Chen, S.J.; Jin, H.C.; Wang, X.; Wang, L.J. Klotho, an anti-senescence related gene, is frequently inactivated through promoter hypermethylation in colorectal cancer. Tumour Biol. 2011, 32, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, Y.; Fan, Z.; Ji, G.; Wang, M.; Lin, J.; Huang, S.; Meltzer, S.J. Klotho: A tumor suppressor and modulator of the Wnt/β-catenin pathway in human hepatocellular carcinoma. Lab Investig. 2016, 96, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Wolf, I.; Levanon-Cohen, S.; Bose, S.; Ligumsky, H.; Sredni, B.; Kanety, H.; Kuro-o, M.; Karlan, B.; Kaufman, B.; Koeffler, H.P.; et al. Klotho: A tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 2008, 27, 7094–7105. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Zhang, Q.; Yang, J.; Lin, W.; Li, Y.; Chen, F.; Cao, W. TGFβ-incurred epigenetic aberrations of miRNA and DNA methyltransferase suppress Klotho and potentiate renal fibrosis. Biochim. Biophys. Acta 2017, 1864, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Sun, Y.; He, P. Bone Morphogenetic Protein-7 Inhibits EMT-Associated Genes in Breast Cancer. Cell Physiol. Biochem. 2015, 37, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Yamashita, H.; Sampath, T.K.; Reddi, A.H.; Estevez, M.; Riddle, D.L.; Ichijo, H.; Heldin, C.H.; Miyazono, K. Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J. Biol. Chem. 1994, 269, 16985–16988. [Google Scholar] [PubMed]
- Derynck, R.; Feng, X.H. TGF-β receptor signaling. Biochim. Biophys. Acta 1997, 1333, F105–F150. [Google Scholar] [CrossRef]
- Bach, D.H.; Park, H.J.; Lee, S.K. The Dual Role of Bone Morphogenetic Proteins in Cancer. Mol. Ther. Oncolytics 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zabkiewicz, C.; Resaul, J.; Hargest, R.; Jiang, W.G.; Ye, L. Bone morphogenetic proteins, breast cancer, and bone metastases: Striking the right balance. Endocr.-Relat. Cancer 2017, 24, R349–R366. [Google Scholar] [CrossRef] [PubMed]
- Tate, C.M.; Pallini, R.; Ricci-Vitiani, L.; Dowless, M.; Shiyanova, T.; D'Alessandris, G.Q.; Morgante, L.; Giannetti, S.; Larocca, L.M.; di Martino, S.; et al. A BMP7 variant inhibits the tumorigenic potential of glioblastoma stem-like cells. Cell Death Differ. 2012, 19, 1644–1654. [Google Scholar] [CrossRef] [PubMed]
- Buijs, J.T.; Rentsch, C.A.; van der Horst, G.; van Overveld, P.G.; Wetterwald, A.; Schwaninger, R.; Henriquez, N.V.; Ten Dijke, P.; Borovecki, F.; Markwalder, R.; et al. BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am. J. Pathol. 2007, 171, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Buijs, J.T.; Henriquez, N.V.; van Overveld, P.G.; van der Horst, G.; Que, I.; Schwaninger, R.; Rentsch, C.; Ten Dijke, P.; Cleton-Jansen, A.M.; Driouch, K.; et al. Bone morphogenetic protein 7 in the development and treatment of bone metastases from breast cancer. Cancer Res. 2007, 67, 8742–8751. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Sun, Y.; He, P. MicroRNA-137 inhibits BMP7 to enhance the epithelial-mesenchymal transition of breast cancer cells. Oncotarget 2017, 8, 18348–18358. [Google Scholar] [CrossRef] [PubMed]
- Dogar, A.M.; Towbin, H.; Hall, J. Suppression of latent transforming growth factor (TGF)-β1 restores growth inhibitory TGF-β signaling through microRNAs. J. Biol. Chem. 2011, 286, 16447–16458. [Google Scholar] [CrossRef] [PubMed]
- Deheuninck, J.; Luo, K. Ski and SnoN, potent negative regulators of TGF-β signaling. Cell Res. 2009, 19, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yin, A.; Zhao, F.; Zhang, W.; Lv, J.; Lv, J. SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor β signaling. Proc. Natl. Acad. Sci. USA 1999, 96, 12442–12447. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Krawitz, A.R.; Chai, J.; Li, W.; Zhang, F.; Luo, K.; Shi, Y. Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: Insights on Ski-mediated repression of TGF-β signaling. Cell 2002, 111, 357–367. [Google Scholar] [CrossRef]
- Fukasawa, H.; Yamamoto, T.; Togawa, A.; Ohashi, N.; Fujigaki, Y.; Oda, T.; Uchida, C.; Kitagawa, K.; Hattori, T.; Suzuki, S.; et al. Ubiquitin-dependent degradation of SnoN and Ski is increased in renal fibrosis induced by obstructive injury. Kidney Int. 2006, 69, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Zhang, J.; Tan, X.; Zhang, X.; Yang, J.; Liu, Y. Downregulation of SnoN expression in obstructive nephropathy is mediated by an enhanced ubiquitin-dependent degradation. J. Am. Soc. Nephrol. 2006, 17, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Heider, T.R.; Lyman, S.; Schoonhoven, R.; Behrns, K.E. Ski promotes tumor growth through abrogation of transforming growth factor-β signaling in pancreatic cancer. Ann. Surg. 2007, 246, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Villanacci, V.; Bellone, G.; Battaglia, E.; Rossi, E.; Carbone, A.; Prati, A.; Verna, C.; Niola, P.; Morelli, A.; Grassini, M.; et al. Ski/SnoN expression in the sequence metaplasia-dysplasia-adenocarcinoma of Barrett’s esophagus. Hum. Pathol. 2008, 39, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, T.; Dong, H.D.; Xu, M.; Maekawa, T.; Ishii, S. The sno gene, which encodes a component of the histone deacetylase complex, acts as a tumor suppressor in mice. EMBO J. 2000, 19, 2280–2291. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, T.; Nomura, T.; Colmenares, C.; Ohira, M.; Nakagawara, A.; Ishii, S. Increased susceptibility to tumorigenesis of ski-deficient heterozygous mice. Oncogene 2001, 20, 8100–8108. [Google Scholar] [CrossRef] [PubMed]
- Onichtchouk, D.; Chen, Y.G.; Dosch, R.; Gawantka, V.; Delius, H.; Massague, J.; Niehrs, C. Silencing of TGF-β signalling by the pseudoreceptor BAMBI. Nature 1999, 401, 480–485. [Google Scholar] [PubMed]
- Sekiya, T.; Adachi, S.; Kohu, K.; Yamada, T.; Higuchi, O.; Furukawa, Y.; Nakamura, Y.; Nakamura, T.; Tashiro, K.; Kuhara, S.; et al. Identification of BMP and activin membrane-bound inhibitor (BAMBI), an inhibitor of transforming growth factor-β signaling, as a target of the β-catenin pathway in colorectal tumor cells. J. Biol. Chem. 2004, 279, 6840–6846. [Google Scholar] [CrossRef] [PubMed]
- Togo, N.; Ohwada, S.; Sakurai, S.; Toya, H.; Sakamoto, I.; Yamada, T.; Nakano, T.; Muroya, K.; Takeyoshi, I.; Nakajima, T.; et al. Prognostic significance of BMP and activin membrane-bound inhibitor in colorectal cancer. World J. Gastroenterol. 2008, 14, 4880–4888. [Google Scholar] [CrossRef] [PubMed]
- Fritzmann, J.; Morkel, M.; Besser, D.; Budczies, J.; Kosel, F.; Brembeck, F.H.; Stein, U.; Fichtner, I.; Schlag, P.M.; Birchmeier, W. A colorectal cancer expression profile that includes transforming growth factor β inhibitor BAMBI predicts metastatic potential. Gastroenterology 2009, 137, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chai, H. Inhibition of BAMBI reduces the viability and motility of colon cancer via activating TGF-β/Smad pathway in vitro and in vivo. Oncol. Lett. 2017, 14, 4793–4799. [Google Scholar] [CrossRef] [PubMed]
- Marwitz, S.; Depner, S.; Dvornikov, D.; Merkle, R.; Szczygiel, M.; Muller-Decker, K.; Lucarelli, P.; Wasch, M.; Mairbaurl, H.; Rabe, K.F.; et al. Downregulation of the TGFβ Pseudoreceptor BAMBI in Non-Small Cell Lung Cancer Enhances TGFβ Signaling and Invasion. Cancer Res. 2016, 76, 3785–3801. [Google Scholar] [CrossRef] [PubMed]
- Khin, S.S.; Kitazawa, R.; Win, N.; Aye, T.T.; Mori, K.; Kondo, T.; Kitazawa, S. BAMBI gene is epigenetically silenced in subset of high-grade bladder cancer. Int. J. Cancer 2009, 125, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Sebens, S.; Giehl, K.; Helm, O.; Groth, S.; Fandrich, F.; Rocken, C.; Sipos, B.; Lehnert, H.; Gieseler, F. Rac1b negatively regulates TGF-β1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signaling. Oncotarget 2014, 5, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Witte, D.; Otterbein, H.; Forster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-β1-induced MKK6-p38 and MEK-ERK signaling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Stallings-Mann, M.L.; Waldmann, J.; Zhang, Y.; Miller, E.; Gauthier, M.L.; Visscher, D.W.; Downey, G.P.; Radisky, E.S.; Fields, A.P.; Radisky, D.C. Matrix metalloproteinase induction of Rac1b, a key effector of lung cancer progression. Sci. Transl. Med. 2012, 4, 142ra195. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Gifford, C.C.; Samarakoon, R.; Higgins, P.J. Deregulation of Negative Controls on TGF-β1 Signaling in Tumor Progression. Cancers 2018, 10, 159. https://doi.org/10.3390/cancers10060159
Tang J, Gifford CC, Samarakoon R, Higgins PJ. Deregulation of Negative Controls on TGF-β1 Signaling in Tumor Progression. Cancers. 2018; 10(6):159. https://doi.org/10.3390/cancers10060159
Chicago/Turabian StyleTang, Jiaqi, Cody C. Gifford, Rohan Samarakoon, and Paul J. Higgins. 2018. "Deregulation of Negative Controls on TGF-β1 Signaling in Tumor Progression" Cancers 10, no. 6: 159. https://doi.org/10.3390/cancers10060159
APA StyleTang, J., Gifford, C. C., Samarakoon, R., & Higgins, P. J. (2018). Deregulation of Negative Controls on TGF-β1 Signaling in Tumor Progression. Cancers, 10(6), 159. https://doi.org/10.3390/cancers10060159