Good Guy or Bad Guy? The Duality of Wild-Type p53 in Hormone-Dependent Breast Cancer Origin, Treatment, and Recurrence


Abstract
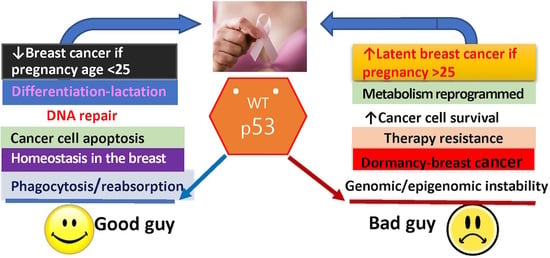
:
1. Introduction
1.1. Overview of Breast Cancer and Pregnancy
1.2. p53 in the Differentiation of the Breast and in Breast Cancer
2. Female Hormones, Pregnancy, and Prevention/Promotion of Breast Cancer
3. Differentiation in the Mammary Gland during Pregnancy and the Origin of Breast Cancer
4. Female Hormones, Pregnancy, and Prevention of Breast Cancer
4.1. Can Estrogen and Progesterone Mimic Pregnancy-Induced Breast Cancer Protection?
4.2. Can hCG Mimic Pregnancy-Induced Breast Cancer Protection?
5. p53 in Pregnancy: Cancer Suppressor Protein and Potent Protector again Latent Breast Cancer
5.1. p53: Looking beyond the Guardian of the Genome
5.2. A Role for p53 in Breast Cancer Origin
5.3. Hormonal Activation of P19ARF–p53 in Mammary Development
5.4. Lessons from the p53-Null Mammary Gland Transplantations
5.5. p53 and Mammary Stem Cell Proliferation
5.6. p53 and Mammary Cancer Stem Cell Theory
5.7. p53 and Methylation
6. The Connection between p53 Status and Responsiveness to Female Hormones in Breast Cancer
6.1. “Paracrine to Autocrine” Hormonal Response in Normal to Breast Cancer Transition
6.2. Replacement of Female Hormones and Breast Cancer Risk
6.3. p53 Status in Hormone-Responsive Breast Cancer
6.4. Molecular Basis for p53 and ERα Association in Resistance and Recurrence in Breast Cancer
6.5. p53 and ERβ Association in Breast Cancer Prevention
6.6. Hypothesis of p53 Activation Changing Metabolism and Breast Cancer Survival
6.7. Problems Raised with p53-Based Treatments in ERα-positive Breast Cancer
7. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Zheng, R.; Zeng, H.; Zhang, S.; Chen, W. Estimates of cancer incidence and mortality in China, 2013. Chin. J. Cancer 2017, 36, 66. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual report to the nation on the status of cancer, 1975–2014, featuring survival. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, O.; Bray, F.; Coleman, M.P.; Vanderpuye, V.; Eniu, A.; Kotha, S.R.; Sarker, M.; Huong, T.T.; Allemani, C.; Dvaladze, A.; et al. The global burden of women’s cancers: A grand challenge in global health. Lancet 2017, 389, 847–860. [Google Scholar] [CrossRef]
- Benz, C.C. Impact of aging on the biology of breast cancer. Crit. Rev. Oncol. Hematol. 2008, 66, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Beatson, G.T. On the treatment of inoperable cases of carcinoma of the mamma: Suggestions for a new method of treatment, with illustrative cases. Lancet 1896, 148, 162–165. [Google Scholar] [CrossRef]
- Troisi, R.; Bjorge, T.; Gissler, M.; Grotmol, T.; Kitahara, C.M.; Myrtveit Saether, S.M.; Ording, A.G.; Skold, C.; Sorensen, H.T.; Trabert, B.; et al. The role of pregnancy, perinatal factors and hormones in maternal cancer risk: A review of the evidence. J. Intern. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Carioli, G.; Malvezzi, M.; Rodriguez, T.; Bertuccio, P.; Negri, E.; La Vecchia, C. Trends and predictions to 2020 in breast cancer mortality in Europe. Breast 2017, 36, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Strasser-Weippl, K.; Li, J.J.; St Louis, J.; Finkelstein, D.M.; Yu, K.D.; Chen, W.Q.; Shao, Z.M.; Goss, P.E. Breast cancer in China. Lancet Oncol. 2014, 15, e279–e289. [Google Scholar] [CrossRef]
- Sivaraman, L.; Conneely, O.M.; Medina, D.; O’Malley, B.W. P53 is a potential mediator of pregnancy and hormone-induced resistance to mammary carcinogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 12379–12384. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. P53 induces differentiation of mouse embryonic stem cells by suppressing nanog expression. Nat. Cell Biol. 2005, 7, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.; Kittrell, F.S. P53 function is required for hormone-mediated protection of mouse mammary tumorigenesis. Cancer Res. 2003, 63, 6140–6143. [Google Scholar] [PubMed]
- Jerry, D.J.; Kittrell, F.S.; Kuperwasser, C.; Laucirica, R.; Dickinson, E.S.; Bonilla, P.J.; Butel, J.S.; Medina, D. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene 2000, 19, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, L.; Stephens, L.C.; Markaverich, B.M.; Clark, J.A.; Krnacik, S.; Conneely, O.M.; O’Malley, B.W.; Medina, D. Hormone-induced refractoriness to mammary carcinogenesis in wistar-furth rats. Carcinogenesis 1998, 19, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Swanson, S.M.; Whitaker, L.M.; Stockard, C.R.; Myers, R.B.; Oelschlager, D.; Grizzle, W.E.; Juliana, M.M.; Grubbs, C.J. Hormone levels and mammary epithelial cell proliferation in rats treated with a regimen of estradiol and progesterone that mimics the preventive effect of pregnancy against mammary cancer. Anticancer Res. 1997, 17, 4639–4645. [Google Scholar] [PubMed]
- Grubbs, C.J.; Juliana, M.M.; Whitaker, L.M. Short-term hormone treatment as a chemopreventive method against mammary cancer initiation in rats. Anticancer Res. 1988, 8, 113–117. [Google Scholar] [PubMed]
- Huggins, C.; Moon, R.C.; Morii, S. Extinction of experimental mammary cancer. I. Estradiol-17β and progesterone. Proc. Natl. Acad. Sci. USA 1962, 48, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.; Peterson, L.E.; Moraes, R.; Gay, J. Short-term exposure to estrogen and progesterone induces partial protection against n-nitroso-n-methylurea-induced mammary tumorigenesis in wistar–furth rats. Cancer Lett. 2001, 169, 1–6. [Google Scholar] [CrossRef]
- Russo, I.H.; Russo, J. Role of hormones in mammary cancer initiation and progression. J. Mammary Gland Biol. Neoplasia 1998, 3, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.H.; Russo, J. Hormonal approach to breast cancer prevention. J. Cell. Biochem. Suppl. 2000, 34, 1–6. [Google Scholar] [CrossRef]
- Horn, J.; Vatten, L.J. Reproductive and hormonal risk factors of breast cancer: A historical perspective. Int. J. Womens Health 2017, 9, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, S.; Teymourzadeh, A.; Farahmand, L.; Majidzadeh-A, K. Challenges of endocrine therapy in breast cancer. In Cancer Genetics and Psychotherapy; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Palmieri, C.; Patten, D.K.; Januszewski, A.; Zucchini, G.; Howell, S.J. Breast cancer: Current and future endocrine therapies. Mol. Cell. Endocrinol. 2014, 382, 695–723. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group. Aromatase inhibitors versus tamoxifen in early breast cancer: Patient-level meta-analysis of the randomised trials. Lancet 2015, 386, 1341–1352. [Google Scholar]
- Albrektsen, G.; Heuch, I.; Hansen, S.; Kvale, G. Breast cancer risk by age at birth, time since birth and time intervals between births: Exploring interaction effects. Br. J. Cancer 2005, 92, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Lambe, M.; Hsieh, C.; Trichopoulos, D.; Ekbom, A.; Pavia, M.; Adami, H.O. Transient increase in the risk of breast cancer after giving birth. N. Engl. J. Med. 1994, 331, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.; Pavia, M.; Lambe, M.; Lan, S.J.; Colditz, G.A.; Ekbom, A.; Adami, H.O.; Trichopoulos, D.; Willett, W.C. Dual effect of parity on breast cancer risk. Eur. J. Cancer 1994, 30A, 969–973. [Google Scholar] [CrossRef]
- Barton, M.; Santucci-Pereira, J.; Russo, J. Molecular pathways involved in pregnancy-induced prevention against breast cancer. Front. Endocrinol. 2014, 5, 213. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Pfeiffer, R.M.; Dores, G.M.; Sherman, M.E. Comparison of age distribution patterns for different histopathologic types of breast carcinoma. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Rosner, B.; Colditz, G.A.; Willett, W.C. Reproductive risk factors in a prospective study of breast cancer: The nurses’ health study. Am. J. Epidemiol. 1994, 139, 819–835. [Google Scholar] [CrossRef] [PubMed]
- MacMahon, B.; Cole, P.; Lin, T.M.; Lowe, C.R.; Mirra, A.P.; Ravnihar, B.; Salber, E.J.; Valaoras, V.G.; Yuasa, S. Age at first birth and breast cancer risk. Bull. World Health Organ. 1970, 43, 209–221. [Google Scholar] [PubMed]
- Mustacchi, P. Ramazzini and rigoni-stern on parity and breast cancer. Clinical impression and statistical corroboration. Arch. Intern. Med. 1961, 108, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Russo, J. Significance of rat mammary tumors for human risk assessment. Toxicol. Pathol. 2015, 43, 145–170. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.K.; Pazik, J.E.; Dao, T.L. Prevention of mammary carcinogenesis in rats by pregnancy: Effect of full-term and interrupted pregnancy. Br. J. Cancer 1988, 57, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, C.J.; Farnell, D.R.; Hill, D.L.; McDonough, K.C. Chemoprevention of n-nitroso-n-methylurea-induced mammary cancers by pretreatment with 17 β-estradiol and progesterone. J. Natl. Cancer Inst. 1985, 74, 927–931. [Google Scholar] [PubMed]
- Russo, J.; Wilgus, G.; Tait, L.; Russo, I.H. Influence of age and parity on the susceptibility of rat mammary gland epithelial cells in primary cultures to 7,12-dimethylbenz(a)anthracene. In Vitro 1981, 17, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Swanson, S.M.; Guzman, R.C.; Collins, G.; Tafoya, P.; Thordarson, G.; Talamantes, F.; Nandi, S. Refractoriness to mammary carcinogenesis in the parous mouse is reversible by hormonal stimulation induced by pituitary isografts. Cancer Lett. 1995, 90, 171–181. [Google Scholar] [CrossRef]
- Russo, I.H.; Russo, J. Mammary gland neoplasia in long-term rodent studies. Environ. Health Perspect. 1996, 104, 938–967. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Hu, Y.F.; Silva, I.D.; Russo, I.H. Cancer risk related to mammary gland structure and development. Microsc. Res. Tech. 2001, 52, 204–223. [Google Scholar] [CrossRef]
- Russo, J.; Russo, I.H. Hormonally induced differentiation: A novel approach to breast cancer prevention. J. Cell. Biochem. Suppl. 1995, 22, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Russo, I.H. Influence of differentiation and cell kinetics on the susceptibility of the rat mammary gland to carcinogenesis. Cancer Res. 1980, 40, 2677–2687. [Google Scholar] [PubMed]
- Russo, J.; Russo, I.H. Toward a physiological approach to breast cancer prevention. Cancer Epidemiol. Biomark. Prev. 1994, 3, 353–364. [Google Scholar]
- Alvarado, A.; Lopes, A.C.; Faustino-Rocha, A.I.; Cabrita, A.M.S.; Ferreira, R.; Oliveira, P.A.; Colaco, B. Prognostic factors in mnu and dmba-induced mammary tumors in female rats. Pathol. Res. Pract. 2017, 213, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Santucci-Pereira, J.; de Cicco, R.L.; Sheriff, F.; Russo, P.A.; Peri, S.; Slifker, M.; Ross, E.; Mello, M.L.; Vidal, B.C.; et al. Pregnancy-induced chromatin remodeling in the breast of postmenopausal women. Int. J. Cancer 2012, 131, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Guzman, R.C.; Yang, J.; Rajkumar, L.; Thordarson, G.; Chen, X.; Nandi, S. Hormonal prevention of breast cancer: Mimicking the protective effect of pregnancy. Proc. Natl. Acad. Sci. USA 1999, 96, 2520–2525. [Google Scholar] [CrossRef] [PubMed]
- Beral, V. Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the million women study. Lancet 2003, 362, 419–427. [Google Scholar] [CrossRef]
- Jensen, E.V.; Jordan, V.C. The estrogen receptor: A model for molecular medicine. Clin. Cancer Res. 2003, 9, 1980–1989. [Google Scholar] [PubMed]
- Ali, S.; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Buluwela, L.; Coombes, R.C. Antiestrogens and their therapeutic applications in breast cancer and other diseases. Annu. Rev. Med. 2011, 62, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Medina, D. Mammary developmental fate and breast cancer risk. Endocr. Relat. Cancer 2005, 12, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Wilgus, G.; Russo, I.H. Susceptibility of the mammary gland to carcinogenesis: I differentiation of the mammary gland as determinant of tumor incidence and type of lesion. Am. J. Pathol. 1979, 96, 721–736. [Google Scholar] [PubMed]
- Ginger, M.R.; Gonzalez-Rimbau, M.F.; Gay, J.P.; Rosen, J.M. Persistent changes in gene expression induced by estrogen and progesterone in the rat mammary gland. Mol. Endocrinol. 2001, 15, 1993–2009. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yoshizawa, K.; Nandi, S.; Tsubura, A. Protective effects of pregnancy and lactation against n-methyl-n-nitrosourea-induced mammary carcinomas in female lewis rats. Carcinogenesis 1999, 20, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Schuler-Toprak, S.; Treeck, O.; Ortmann, O. Human chorionic gonadotropin and breast cancer. Int. J. Mol. Sci. 2017, 18, 1587. [Google Scholar] [CrossRef] [PubMed]
- Santucci-Pereira, J.; George, C.; Armiss, D.; Russo, I.H.; Vanegas, J.E.; Sheriff, F.; de Cicco, R.L.; Su, Y.; Russo, P.A.; Bidinotto, L.T.; et al. Mimicking pregnancy as a strategy for breast cancer prevention. Breast Cancer Manag. 2013, 2, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Janssens, J.P.; Russo, J.; Russo, I.; Michiels, L.; Donders, G.; Verjans, M.; Riphagen, I.; Van den Bossche, T.; Deleu, M.; Sieprath, P. Human chorionic gonadotropin (hcg) and prevention of breast cancer. Mol. Cell. Endocrinol. 2007, 269, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Fortner, R.T.; Schock, H.; Kaaks, R.; Lehtinen, M.; Pukkala, E.; Lakso, H.A.; Tanner, M.; Kallio, R.; Joensuu, H.; Korpela, J.; et al. Human chorionic gonadotropin does not correlate with risk for maternal breast cancer: Results from the finnish maternity cohort. Cancer Res. 2017, 77, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H. P53: Death star. Cell 2000, 103, 691–694. [Google Scholar] [CrossRef]
- Vousden, K.H.; Vande Woude, G.F. The ins and outs of p53. Nat. Cell Biol. 2000, 2, E178–E180. [Google Scholar] [CrossRef] [PubMed]
- Rotter, V.; Foord, O.; Navot, N. In search of the functions of normal p53 protein. Trends Cell Biol. 1993, 3, 46–49. [Google Scholar] [CrossRef]
- Tovy, A.; Spiro, A.; McCarthy, R.; Shipony, Z.; Aylon, Y.; Allton, K.; Ainbinder, E.; Furth, N.; Tanay, A.; Barton, M.; et al. P53 is essential for DNA methylation homeostasis in naive embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev. 2017, 31, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. The paradox of p53: What, how, and why? Cold Spring Harb. Perspect. Med. 2016, 6, a026328. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.; Kittrell, F.S.; Shepard, A.; Contreras, A.; Rosen, J.M.; Lydon, J. Hormone dependence in premalignant mammary progression. Cancer Res. 2003, 63, 1067–1072. [Google Scholar] [PubMed]
- Park, I.Y.; Sohn, B.H.; Choo, J.H.; Joe, C.O.; Seong, J.K.; Lee, Y.I.; Chung, J.H. Deregulation of DNA methyltransferases and loss of parental methylation at the insulin-like growth factor ii (igf2)/h19 loci in p53 knockout mice prior to tumor development. J. Cell. Biochem. 2005, 94, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The p53 protein plays a central role in the mechanism of action of epigentic drugs that alter the methylation of cytosine residues in DNA. Oncotarget 2017, 8, 7228–7230. [Google Scholar] [CrossRef] [PubMed]
- West, L.E.; Gozani, O. Regulation of p53 function by lysine methylation. Epigenomics 2011, 3, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. P53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M. P53 and autophagy in cancer: Guardian of the genome meets guardian of the proteome. Eur. J. Cancer 2011, 47, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.S.; Ramos, H.; Soares, J.; Saraiva, L. P53 and glucose metabolism: An orchestra to be directed in cancer therapy. Pharmacol. Res. 2018, 131, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Itahana, K. Emerging roles of p53 family members in glucose metabolism. Int. J. Mol. Sci. 2018, 19, 776. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. P53 as a regulator of lipid metabolism in cancer. Int. J. Mol. Sci. 2016, 17, 2074. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P. P53 in metabolism, aging and cancer. Ann. Dermatol. Venereol. 2014, 141, S200–S201. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Yang, X. P53 and regulation of tumor metabolism. J. Carcinog. 2013, 12, 21. [Google Scholar] [PubMed]
- Sen, N.; Satija, Y.K.; Das, S. P53 and metabolism: Old player in a new game. Transcription 2012, 3, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. P53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.; Hanisch, J.J.; Choi, J.W.; Hwang, P.M. How does p53 regulate mitochondrial respiration? IUBMB Life 2007, 59, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A pivotal role for p53: Balancing aerobic respiration and glycolysis. J. Bioenerg. Biomembr. 2007, 39, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. P53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The role of p53 in glucose metabolism. Curr. Opin. Cell Biol. 2010, 22, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Lin, Y. P53 switches off pluripotency on differentiation. Stem Cell Res. Ther. 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Tomaszowski, K.H.; Luzwick, J.W.; Park, S.; Li, J.; Murphy, M.; Schlacher, K. P53 orchestrates DNA replication restart homeostasis by suppressing mutagenic rad52 and poltheta pathways. eLife 2018, 7, e31723. [Google Scholar] [CrossRef] [PubMed]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Legrand, A.J.; Fletcher, S.C.; Dianov, G.L. P53 coordinates base excision repair to prevent genomic instability. Nucleic Acids Res. 2016, 44, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.Q.; Lowman, X.H.; Reid, M.A.; Mendez-Dorantes, C.; Pan, M.; Yang, Y.; Kong, M. Tumor-associated mutant p53 promotes cancer cell survival upon glutamine deprivation through p21 induction. Oncogene 2017, 36, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- McGowan, E.M.; Alling, N.; Jackson, E.A.; Yagoub, D.; Haass, N.K.; Allen, J.D.; Martinello-Wilks, R. Evaluation of cell cycle arrest in estrogen responsive mcf-7 breast cancer cells: Pitfalls of the mts assay. PLoS ONE 2011, 6, e20623. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.B.; Lu, M.; Eilers, G.; Li, H.; Ding, J.; Meng, X.; Wu, Y.; He, Q.; Sheng, Q.; Zhou, H.M.; et al. Co-targeting of fak and mdm2 triggers additive anti-proliferative effects in mesothelioma via a coordinated reactivation of p53. Br. J. Cancer 2016, 115, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.T.; Shin, H.; Westerling, T.; Liu, X.S.; Brown, M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 18060–18065. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintas-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. P53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Hatoum, D.; Yagoub, D.; Ahadi, A.; Nassif, N.T.; McGowan, E.M. Annexin/s100a protein family regulation through p14arf-p53 activation: A role in cell survival and predicting treatment outcomes in breast cancer. PLoS ONE 2017, 12, e0169925. [Google Scholar] [CrossRef] [PubMed]
- McGowan, E.M.; Tran, N.; Alling, N.; Yagoub, D.; Sedger, L.M.; Martiniello-Wilks, R. P14arf post-transcriptional regulation of nuclear cyclin d1 in mcf-7 breast cancer cells: Discrimination between a good and bad prognosis? PLoS ONE 2012, 7, e42246. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. P53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. P53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009, 4, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Dimri, G.P.; Hara, E.; Itahana, Y.; Zou, Y.; Desprez, P.Y.; Campisi, J. A role for p53 in maintaining and establishing the quiescence growth arrest in human cells. J. Biol. Chem. 2002, 277, 18206–18214. [Google Scholar] [CrossRef] [PubMed]
- Korotchkina, L.G.; Leontieva, O.V.; Bukreeva, E.I.; Demidenko, Z.N.; Gudkov, A.V.; Blagosklonny, M.V. The choice between p53-induced senescence and quiescence is determined in part by the mtor pathway. Aging 2010, 2, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Allred, D.C.; Clark, G.M.; Elledge, R.; Fuqua, S.A.; Brown, R.W.; Chamness, G.C.; Osborne, C.K.; McGuire, W.L. Association of p53 protein expression with tumor cell proliferation rate and clinical outcome in node-negative breast cancer. J. Natl. Cancer Inst. 1993, 85, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.B.; Yin, D.D.; Sun, M.; Kong, R.; Liu, X.H.; You, L.H.; Han, L.; Xia, R.; Wang, K.M.; Yang, J.S.; et al. P53-regulated long non-coding rna tug1 affects cell proliferation in human non-small cell lung cancer, partly through epigenetically regulating hoxb7 expression. Cell Death Dis. 2014, 5, e1243. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Abrams, J. P53: The janus of autophagy? Nat. Cell Biol. 2008, 10, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. Dram, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mtor pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer—An overview and update. Mol. Cell. Endocrinol. 2015, 418 Pt 3, 220–234. [Google Scholar] [CrossRef]
- Gottlieb, E.; Vousden, K.H. P53 regulation of metabolic pathways. Cold Spring Harb. Perspect. Biol. 2010, 2, a001040. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. P53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Walerych, D.; Sal, G.D. Targeting mutant p53 in cancer: A long road to precision therapy. FEBS J. 2017, 284, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Escoll, M.; Gargini, R.; Cuadrado, A.; Anton, I.M.; Wandosell, F. Mutant p53 oncogenic functions in cancer stem cells are regulated by wip through yap/taz. Oncogene 2017, 36, 3515–3527. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Mirza, S.A.; Xu, S.; Schulz-Heddergott, R.; Marchenko, N.D.; Moll, U.M. P53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis. 2017, 8, e2661. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Prives, C. P53: Master of life, death, and the epigenome. Genes Dev. 2017, 31, 955–956. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Pappas, K.; Xu, J.; Zairis, S.; Resnick-Silverman, L.; Abate, F.; Steinbach, N.; Ozturk, S.; Saal, L.H.; Su, T.; Cheung, P.; et al. P53 maintains baseline expression of multiple tumor suppressor genes. Mol. Cancer Res. 2017, 15, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Vousden, K.H. Hitting cancers’ weak spots: Vulnerabilities imposed by p53 mutation. Trends Cell Biol. 2015, 25, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Iwakuma, T. Non-canonical cell death induced by p53. Int. J. Mol. Sci. 2016, 17, 2068. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.; Sivaraman, L.; Hilsenbeck, S.G.; Conneely, O.; Ginger, M.; Rosen, J.; Omalle, B.W. Mechanisms of hormonal prevention of breast cancer. Ann. N. Y. Acad. Sci. 2001, 952, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Ginger, M.R.; Rosen, J.M. Pregnancy-induced changes in cell-fate in the mammary gland. Breast Cancer Res. 2003, 5, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.; Shepard, A.; Kittrell, F.; Mulac-Jericevic, B.; Medina, D.; Said, T.K. P19arf determines the balance between normal cell proliferation rate and apoptosis during mammary gland development. Mol. Biol. Cell 2004, 15, 2302–2311. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16ink4a with retention of p19arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Roa, M.; Malumbres, M. Fueling the cell division cycle. Trends Cell Biol. 2017, 27, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Beatson, G.T. On the treatment of inoperable cases of carcinoma of the mamma: Suggestions for a new method of treatment, with illustrative cases. Trans. Med. Chir. Soc. Edinb. 1896, 15, 153–179. [Google Scholar] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Van de Moosdijk, A.A.; Fu, N.Y.; Rios, A.C.; Visvader, J.E.; van Amerongen, R. Lineage tracing of mammary stem and progenitor cells. Methods Mol. Biol. 2017, 1501, 291–308. [Google Scholar] [PubMed]
- Fu, N.Y.; Rios, A.C.; Pal, B.; Law, C.W.; Jamieson, P.; Liu, R.; Vaillant, F.; Jackling, F.; Liu, K.H.; Smyth, G.K.; et al. Identification of quiescent and spatially restricted mammary stem cells that are hormone responsive. Nat. Cell Biol. 2017, 19, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Stingl, J.; Raouf, A.; Emerman, J.T.; Eaves, C.J. Epithelial progenitors in the normal human mammary gland. J. Mammary Gland Biol. Neoplasia 2005, 10, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009, 138, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Al-Hajj, M.; Abdallah, W.M.; Clarke, M.F.; Wicha, M.S. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003, 36 (Suppl. S1), S59–S72. [Google Scholar] [CrossRef]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Berger, S.L. The interplay between epigenetic changes and the p53 protein in stem cells. Genes Dev. 2017, 31, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Jerry, D.J.; Dunphy, K.A.; Hagen, M.J. Estrogens, regulation of p53 and breast cancer risk: A balancing act. Cell. Mol. Life Sci. 2010, 67, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Leutz, A. Chromatin remodeling in development and differentiation. Curr. Opin. Genet. Dev. 2001, 11, 167–174. [Google Scholar] [CrossRef]
- Clarke, R.B.; Howell, A.; Potten, C.S.; Anderson, E. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Res. 1997, 57, 4987–4991. [Google Scholar] [PubMed]
- Anderson, E. The role of oestrogen and progesterone receptors in human mammary development and tumorigenesis. Breast Cancer Res. 2002, 4, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Anderson, G.; Manson, J.E.; Pettinger, M.; Yasmeen, S.; Lane, D.; Langer, R.D.; Hubbell, F.A.; McTiernan, A.; Hendrix, S.; et al. Estrogen alone in postmenopausal women and breast cancer detection by means of mammography and breast biopsy. J. Clin. Oncol. 2010, 28, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Anderson, G.L. Menopausal hormone therapy and breast cancer mortality: Clinical implications. Ther. Adv. Drug Saf. 2015, 6, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Gurney, E.P.; Nachtigall, M.J.; Nachtigall, L.E.; Naftolin, F. The women’s health initiative trial and related studies: 10 years later: A clinician’s view. J. Steroid Biochem. Mol. Biol. 2014, 142, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Anderson, G.L.; Gass, M.; Lane, D.S.; Aragaki, A.K.; Kuller, L.H.; Manson, J.E.; Stefanick, M.L.; Ockene, J.; Sarto, G.E.; et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010, 304, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Manson, J.E.; Anderson, G.L.; Cauley, J.A.; Aragaki, A.K.; Stefanick, M.L.; Lane, D.S.; Johnson, K.C.; Wactawski-Wende, J.; Chen, C.; et al. Estrogen plus progestin and breast cancer incidence and mortality in the women’s health initiative observational study. J. Natl. Cancer Inst. 2013, 105, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on tp53 mutation patterns and tumor phenotype: Lessons from recent developments in the iarc tp53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Pharoah, P.D.; Day, N.E.; Caldas, C. Somatic mutations in the p53 gene and prognosis in breast cancer: A meta-analysis. Br. J. Cancer 1999, 80, 1968–1973. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Toillon, R.A.; Leclercq, G. P53 and breast cancer, an update. Endocr. Relat. Cancer 2006, 13, 293–325. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Yang, J.; Murphy, R.F.; Agrawal, D.K. Regulation of the p14arf-mdm2-p53 pathway: An overview in breast cancer. Exp. Mol. Pathol. 2006, 81, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Vijayakumaran, R.; Miranda, P.J.; Burgess, A.; Lim, E.; Haupt, Y. The role of mdm2 and mdm4 in breast cancer development and prevention. J. Mol. Cell Biol. 2017, 9, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical overview of mdm2/x-targeted therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakagawara, A. Role of p53 in cell death and human cancers. Cancers (Basel) 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Turpin, E.; Bieche, I.; Bertheau, P.; Plassa, L.F.; Lerebours, F.; de Roquancourt, A.; Olivi, M.; Espie, M.; Marty, M.; Lidereau, R.; et al. Increased incidence of erbb2 overexpression and tp53 mutation in inflammatory breast cancer. Oncogene 2002, 21, 7593–7597. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Paik, A.; Li, J.J. P53 activation in chronic radiation-treated breast cancer cells: Regulation of mdm2/p14arf. Cancer Res. 2004, 64, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Neven, P.; Loibl, S.; Andre, F. Advances in the treatment of advanced oestrogen-receptor-positive breast cancer. Lancet 2017, 389, 2403–2414. [Google Scholar] [CrossRef]
- Konduri, S.D.; Medisetty, R.; Liu, W.; Kaipparettu, B.A.; Srivastava, P.; Brauch, H.; Fritz, P.; Swetzig, W.M.; Gardner, A.E.; Khan, S.A.; et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 15081–15086. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ip, M.M.; Podgorsak, M.B.; Das, G.M. Disruption of estrogen receptor α-p53 interaction in breast tumors: A novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res. Treat. 2009, 115, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Konduri, S.D.; Bansal, S.; Nayak, B.K.; Rajasekaran, S.A.; Karuppayil, S.M.; Rajasekaran, A.K.; Das, G.M. Estrogen receptor-α binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 2006, 281, 9837–9840. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, A.; Konduri, S.D.; Liu, W.; Bansal, S.; Li, F.; Das, G.M. Estrogen receptor α inhibits p53-mediated transcriptional repression: Implications for the regulation of apoptosis. Cancer Res. 2007, 67, 7746–7755. [Google Scholar] [CrossRef] [PubMed]
- Gudas, J.M.; Nguyen, H.; Li, T.; Sadzewicz, L.; Robey, R.; Wosikowski, K.; Cowan, K.H. Drug-resistant breast cancer cells frequently retain expression of a functional wild-type p53 protein. Carcinogenesis 1996, 17, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Holst, F.; Stahl, P.R.; Ruiz, C.; Hellwinkel, O.; Jehan, Z.; Wendland, M.; Lebeau, A.; Terracciano, L.; Al-Kuraya, K.; Janicke, F.; et al. Estrogen receptor α (esr1) gene amplification is frequent in breast cancer. Nat. Genet. 2007, 39, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cuesta, L.; Anaganti, S.; Hainaut, P.; Olivier, M. Estrogen levels act as a rheostat on p53 levels and modulate p53-dependent responses in breast cancer cell lines. Breast Cancer Res. Treat 2011, 125, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.E.; Qian, Y.; Liu, G.; Chen, H.; Chen, X. P53, a target of estrogen receptor (ER) α, modulates DNA damage-induced growth suppression in ER-positive breast cancer cells. J. Biol. Chem. 2012, 287, 30117–30127. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Gustafsson, J.A. The novel estrogen receptor-β subtype: Potential role in the cell- and promoter-specific actions of estrogens and anti-estrogens. FEBS Lett. 1997, 410, 87–90. [Google Scholar] [CrossRef]
- Tremblay, G.B.; Tremblay, A.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Labrie, F.; Giguere, V. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor β. Mol. Endocrinol. 1997, 11, 353–365. [Google Scholar] [PubMed]
- Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor transcription and transactivation: Estrogen receptor α and estrogen receptor β: Regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res. 2000, 2, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaunay, F.; Pettersson, K.; Tujague, M.; Gustafsson, J.A. Functional differences between the amino-terminal domains of estrogen receptors α and β. Mol. Pharmacol. 2000, 58, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Kim, T.H.; Choi, K.C. Functions and physiological roles of two types of estrogen receptors, ERα and ERβ, identified by estrogen receptor knockout mouse. Lab. Anim. Res. 2012, 28, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.C.; Korach, K.S. Oestrogen receptor knockout mice: Roles for oestrogen receptors α and β in reproductive tissues. Reproduction 2003, 125, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gustafsson, J.A. The different roles of er subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Forster, C.; Makela, S.; Warri, A.; Kietz, S.; Becker, D.; Hultenby, K.; Warner, M.; Gustafsson, J.A. Involvement of estrogen receptor β in terminal differentiation of mammary gland epithelium. Proc. Natl. Acad. Sci. USA 2002, 99, 15578–15583. [Google Scholar] [CrossRef] [PubMed]
- Bado, I.; Nikolos, F.; Rajapaksa, G.; Wu, W.; Castaneda, J.; Krishnamurthy, S.; Webb, P.; Gustafsson, J.A.; Thomas, C. Somatic loss of estrogen receptor β and p53 synergize to induce breast tumorigenesis. Breast Cancer Res. 2017, 19, 79. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Katzenellenbogen, B.S. Estrogen receptor-β modulation of the ERα-p53 loop regulating gene expression, proliferation, and apoptosis in breast cancer. Horm. Cancer 2017, 8, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Bado, I.; Pham, E.; Soibam, B.; Nikolos, F.; Gustafsson, J.A.; Thomas, G. Erβ alters the chemosensitivity of luminal breast cancer cells by regulating p53 function. Oncotarget 2018, 9, 22509–22522. [Google Scholar] [CrossRef]
- Goldenberg, I.S. Hormones and breast cancer: Historical perspectives. Surgery 1963, 53, 285–288. [Google Scholar] [PubMed]
- Moulder, D.E.; Hatoum, D.; Tay, E.; Lin, Y.; McGowan, E.M. The roles of p53 in mitochondrial dynamics and cancer metabolism: The pendulum between survival and death in breast cancer? Cancers 2018, accepted. [Google Scholar]
- Cross, B.M.; Breitwieser, G.E.; Reinhardt, T.A.; Rao, R. Cellular calcium dynamics in lactation and breast cancer: From physiology to pathology. Am. J. Physiol. Cell Physiol. 2013, 306, C515–C526. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochimica et Biophysica Acta 2014, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yom, C.K.; Han, W.; Kim, S.W.; Kim, H.S.; Shin, H.C.; Chang, J.N.; Koo, M.; Noh, D.Y.; Moon, B.I. Clinical significance of annexin a1 expression in breast cancer. J. Breast Cancer 2011, 14, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Ang, E.Z.; Nguyen, H.T.; Sim, H.L.; Putti, T.C.; Lim, L.H. Annexin-1 regulates growth arrest induced by high levels of estrogen in mcf-7 breast cancer cells. Mol. Cancer Res. 2009, 7, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, Y.; Edelweiss, M.; Arun, B.; Rosen, D.; Resetkova, E.; Wu, Y.; Liu, J.; Sahin, A.; Albarracin, C.T. Loss of annexin a1 expression in breast cancer progression. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Ganesan, N.; Tachibana, K.; Rajapakshe, K.; Albarracin, C.T.; Gunaratne, P.H.; Coarfa, C.; Bedrosian, I. Annexin a1 preferentially predicts poor prognosis of basal-like breast cancer patients by activating mtor-s6 signaling. PLoS ONE 2015, 10, e0127678. [Google Scholar] [CrossRef] [PubMed]
- Sobral-Leite, M.; Wesseling, J.; Smit, V.T.; Nevanlinna, H.; van Miltenburg, M.H.; Sanders, J.; Hofland, I.; Blows, F.M.; Coulson, P.; Patrycja, G.; et al. Annexin a1 expression in a pooled breast cancer series: Association with tumor subtypes and prognosis. BMC Med. 2015, 13, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, M.; Kumamoto, K.; Saito, M.; Onozawa, H.; Saito, K.; Abe, N.; Ohtake, T.; Takenoshita, S. Upregulated annexin a1 promotes cellular invasion in triple-negative breast cancer. Oncol. Rep. 2015, 33, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.R.; Koltowski, L.; Ownbey, R.T.; Tuszynski, G.P.; Sharma, M.C. Angiogenesis-associated protein annexin ii in breast cancer: Selective expression in invasive breast cancer and contribution to tumor invasion and progression. Exp. Mol. Pathol. 2006, 81, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yuan, J.; Zhang, J.; Tian, R.; Ji, W.; Zhou, Y.; Yang, Y.; Song, W.; Zhang, F.; Niu, R. Anxa2 binds to stat3 and promotes epithelial to mesenchymal transition in breast cancer cells. Oncotarget 2015, 6, 30975–30992. [Google Scholar] [CrossRef] [PubMed]
- Mussunoor, S.; Murray, G.I. The role of annexins in tumour development and progression. J. Pathol. 2008, 216, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhao, J.; Zhang, M. Expression of annexin a2 and its correlation with drug resistance and recurrence of bladder cancer. Technol. Cancer Res. Treat. 2015, 15, NP61–NP68. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, J.K.; Nylandsted, J. S100 and annexin proteins identify cell membrane damage as the achilles heel of metastatic cancer cells. Cell Cycle 2015, 14, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Puzio-Kuter, A.M. The role of p53 in metabolic regulation. Genes Cancer 2011, 2, 385–391. [Google Scholar] [CrossRef] [PubMed]


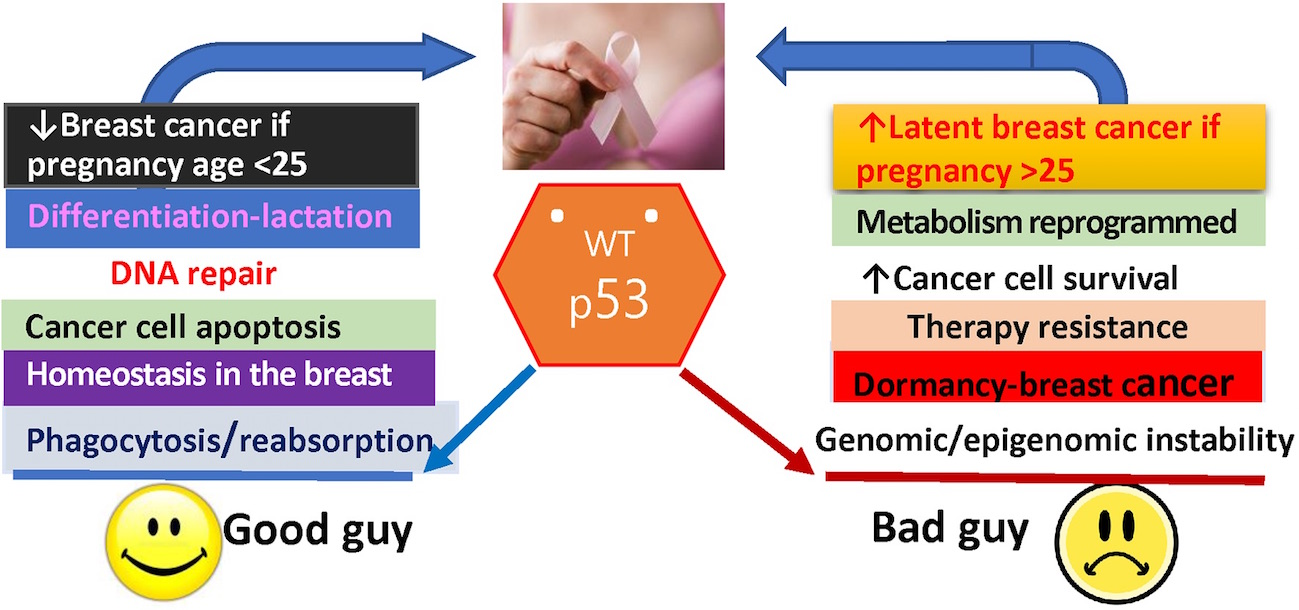{kind=link}
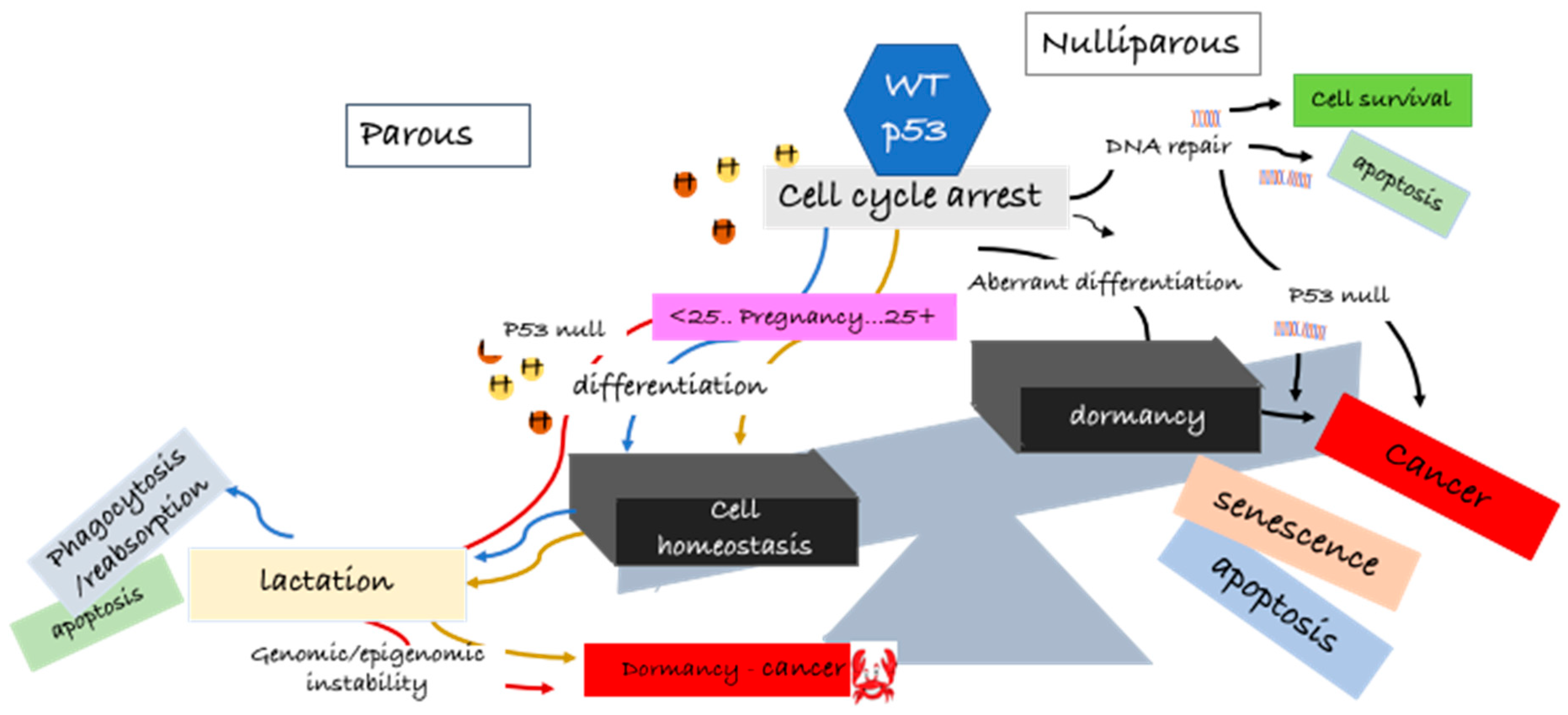{kind=link}
| Function | Summary—Key Regulatory Functions | Reference |
|---|---|---|
| Homeostasis regulators | p53 is a key regulator of replication homeostasis within a DNA restart network and is essential for DNA methylation homeostasis in stem cells. It also plays a key role in the regulation of metabolic homeostasis. The function of p53 in cellular energy homeostasis and metabolism is emerging as a critical factor for tumor suppression. | [62,72,83,84,85] |
| Cell cycle arrest | One of the best-understood function of p53 is to promote cell cycle arrest. Cell cycle arrest by p53 is mainly mediated by the transcriptional activation of p21/WAF1 and is reversible after downregulation of p53. | [72,83,86,87,88,89,90] |
| Apoptosis | It has been confirmed in many studies that induction of apoptotic death in nascent neoplastic cells is the principal mechanism by which p53 suppresses tumor development. p53 induces apoptosis in nontransformed cells mostly by direct transcriptional activation of the pro-apoptotic BH3-only proteins PUMA and (to a lesser extent) NOXA. | [86,87,91,92,93] |
| Cellular senescence | Chronic p53 activation can result in senescence of tumor cells. Senescent cells have unique features, such as large cell size, active autophagy, high lysosomal SA-b-gal activity, and secretion of proinflammatory cytokines. Senescence is a unique state of cell cycle arrest that is highly stable but is not completely irreversible. Through the induction of senescence, p53 promotes and achieves permanent inhibition of cell proliferation. | [86,87,93,94,95,96,97,98,99] |
| Cellular quiescence | p53 is activated during both quiescence and senescence. Evidence suggests that p53 activation contributes to the quiescent growth arrest and is a reversible process. | [100,101,102]. |
| Proliferation/survival | There is a strong direct correlation between accumulation of p53 protein and tumor proliferation rate. Expression of mutant p53 protein was associated with high tumor proliferation rate, early recurrence, and death in breast cancer. Recently, it was noted that p53 can also contribute to cell survival. | [86,90,91,99,103,104] |
| Autophagy | In most cases, p53 positively regulates autophagy in tumor cells by inhibiting mTOR pathways via the activation of AMPK. p53 also promotes autophagy by inducing various autophagy-related genes. Autophagy is considered a tumor suppressive mechanism that removes unfolded proteins, damaged cellular components, and damaged organelles to maintain cellular homeostasis. | [96,105,106,107,108,109,110,111] |
| Metabolism | p53 promotes oxidative phosphorylation and dampens glycolysis in cells; disruption of this balance is associated with mutations in p53 and oncogenic transformation. P53 plays a role in alterations seen in glycolysis, gluconeogenesis, and aerobic respiration. Altered metabolism can contribute to malignant transformation, and cancer cells become dependent on these changes. p53 regulates various metabolic pathways, helping to balance glycolysis and oxidative phosphorylation, limiting the production of reactive oxygen species, and contributing to the ability of cells to adapt to and survive mild metabolic stresses. | [72,77,84,87,88,99,112,113] |
| DNA repair | p53 plays a prominent role as a facilitator of DNA repair by halting the cell cycle to allow time for the repair machinery to restore genomic stability; for example, p53 coordinates DNA base excision repair in the cells, and this mechanism is impaired in p53-inactivated cells. Within a DNA restart network, p53 functions as a keystone regulator in DNA replication homeostasis. | [85,114] |
| Oncogenic functions | p53wt is a tumor suppressor gene; mutations in this gene promote oncogenic capacity. Thus, mutant p53 is an actionable target of clinical antitumor therapies. p53 loss of heterozygosity (LOH ) is a critical prerequisite for missense mutant p53 stabilization and gain of function in vivo. | [92,115,116,117] |
| Epigenomic regulator | p53 is not only a pivotal guardian of genomic stability, but also an epigenetic regulator. Epigenomic regulation is a new function of p53, contributing to its tumor suppressor activity. It is thought that the ability of p53 to maintain DNA methylation balance is an important contributor to its tumor suppressor capacity and that loss of p53 may result in cancer initiation by increasing cellular heterogeneity and epigenetic promiscuity. | [62,93,104,118,119] |
| Regulating multiple tumor suppressor genes | Under normal low-stress conditions, p53wt is capable of maintaining the expression of a group of important tumor suppressor genes at baseline, which could contribute to p53-mediated tumor suppression. p53 mutations, with inactivation of multiple tumor suppressor genes in parallel, could lead to the high frequency of p53 mutations in cancer. | [120] |
| Mutant p53 functions | Unidentified mechanisms by which mutp53 confers oncogenic functions by promoting cancer cell adaptation to metabolic stresses. | [88,92,121] |
| Non-canonical cell death | Transcriptional regulation of downstream targets: caspase-independent apoptosis, autophagy, ferroptosis, mitotic catastrophe, paratosis, pyrotosis, efferocytosis (clearing dead cell debris). | [122] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGowan, E.M.; Lin, Y.; Hatoum, D. Good Guy or Bad Guy? The Duality of Wild-Type p53 in Hormone-Dependent Breast Cancer Origin, Treatment, and Recurrence. Cancers 2018, 10, 172. https://doi.org/10.3390/cancers10060172
McGowan EM, Lin Y, Hatoum D. Good Guy or Bad Guy? The Duality of Wild-Type p53 in Hormone-Dependent Breast Cancer Origin, Treatment, and Recurrence. Cancers. 2018; 10(6):172. https://doi.org/10.3390/cancers10060172
Chicago/Turabian StyleMcGowan, Eileen M., Yiguang Lin, and Diana Hatoum. 2018. "Good Guy or Bad Guy? The Duality of Wild-Type p53 in Hormone-Dependent Breast Cancer Origin, Treatment, and Recurrence" Cancers 10, no. 6: 172. https://doi.org/10.3390/cancers10060172
APA StyleMcGowan, E. M., Lin, Y., & Hatoum, D. (2018). Good Guy or Bad Guy? The Duality of Wild-Type p53 in Hormone-Dependent Breast Cancer Origin, Treatment, and Recurrence. Cancers, 10(6), 172. https://doi.org/10.3390/cancers10060172