The Role of JMY in p53 Regulation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
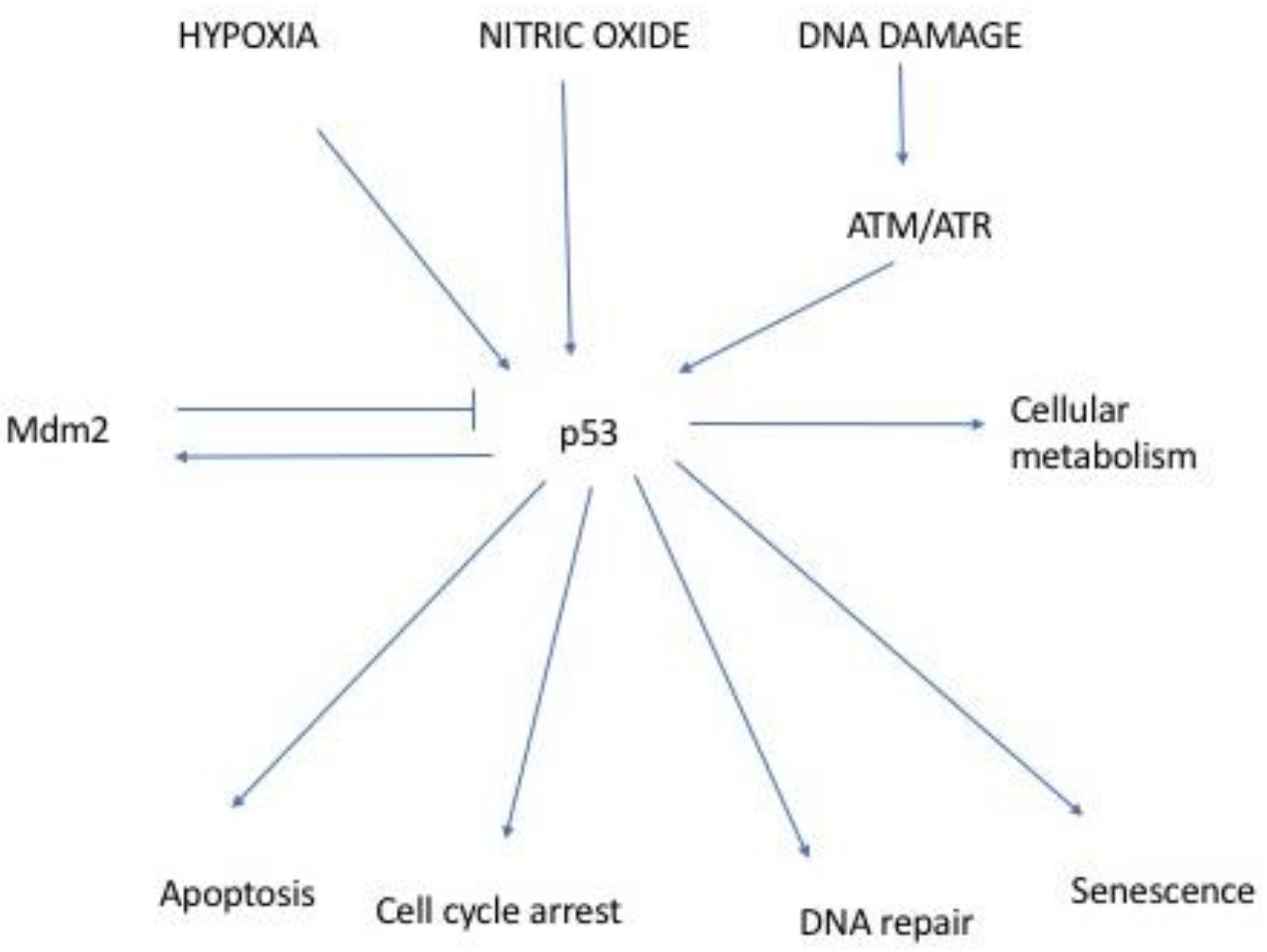
:1. Introduction
2. Identification of JMY
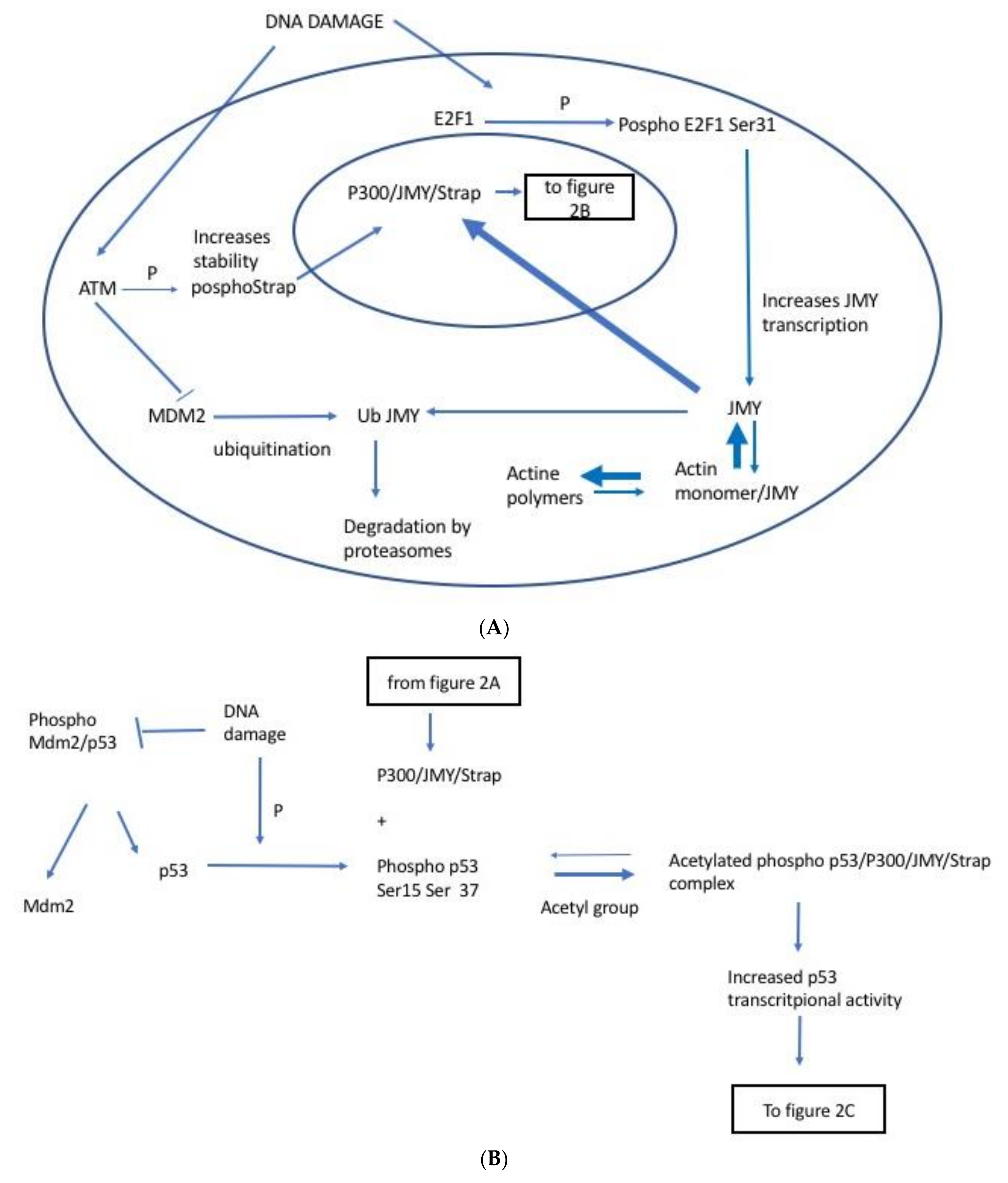
2.1. JMY Regulation by E2F1
2.2. JMY Regulation by Strap
2.3. JMY Regulation by MDM2
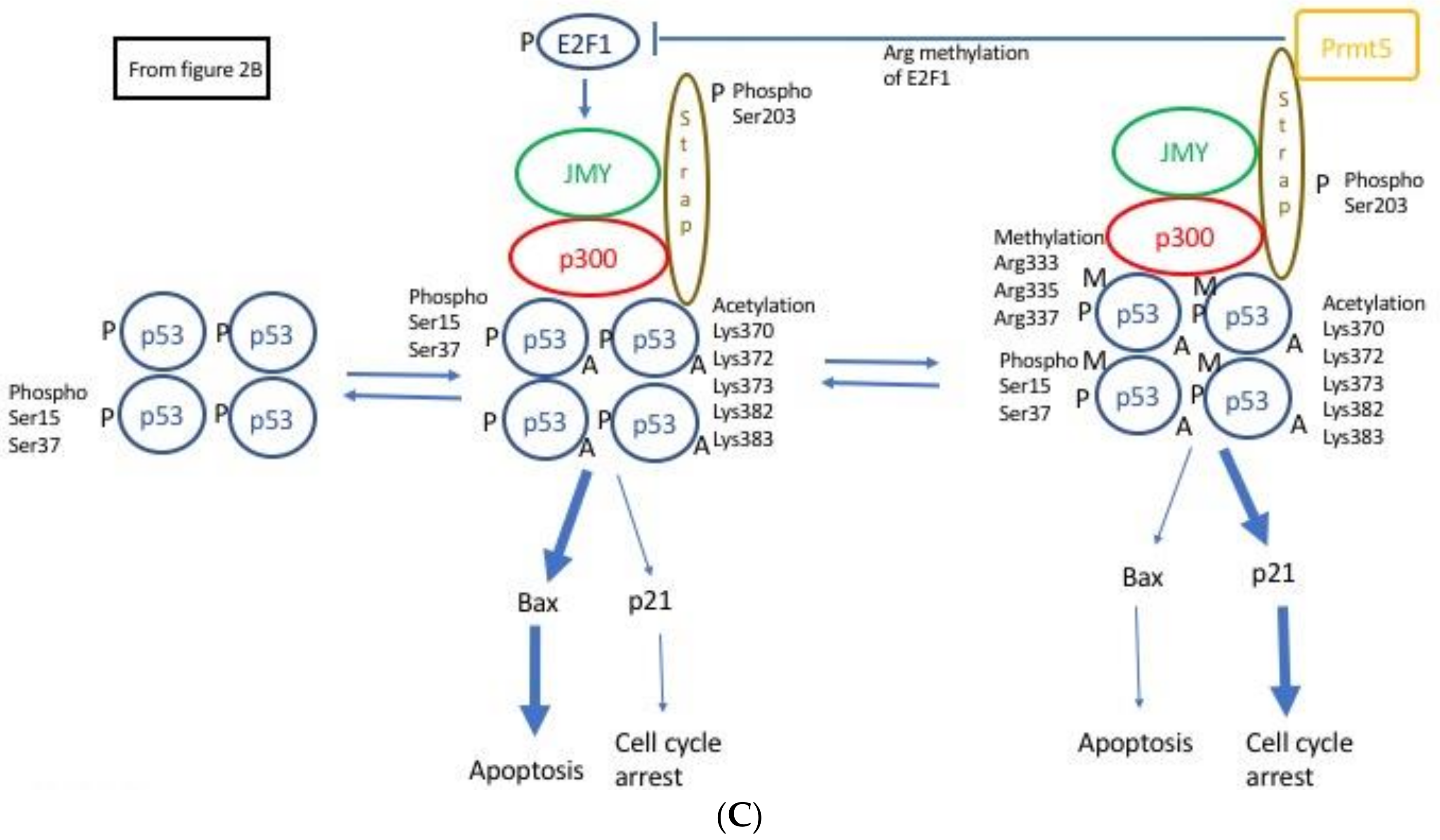
2.4. JMY Increases p53 Dependent Transcription Leading to Selective Increase in Apoptosis
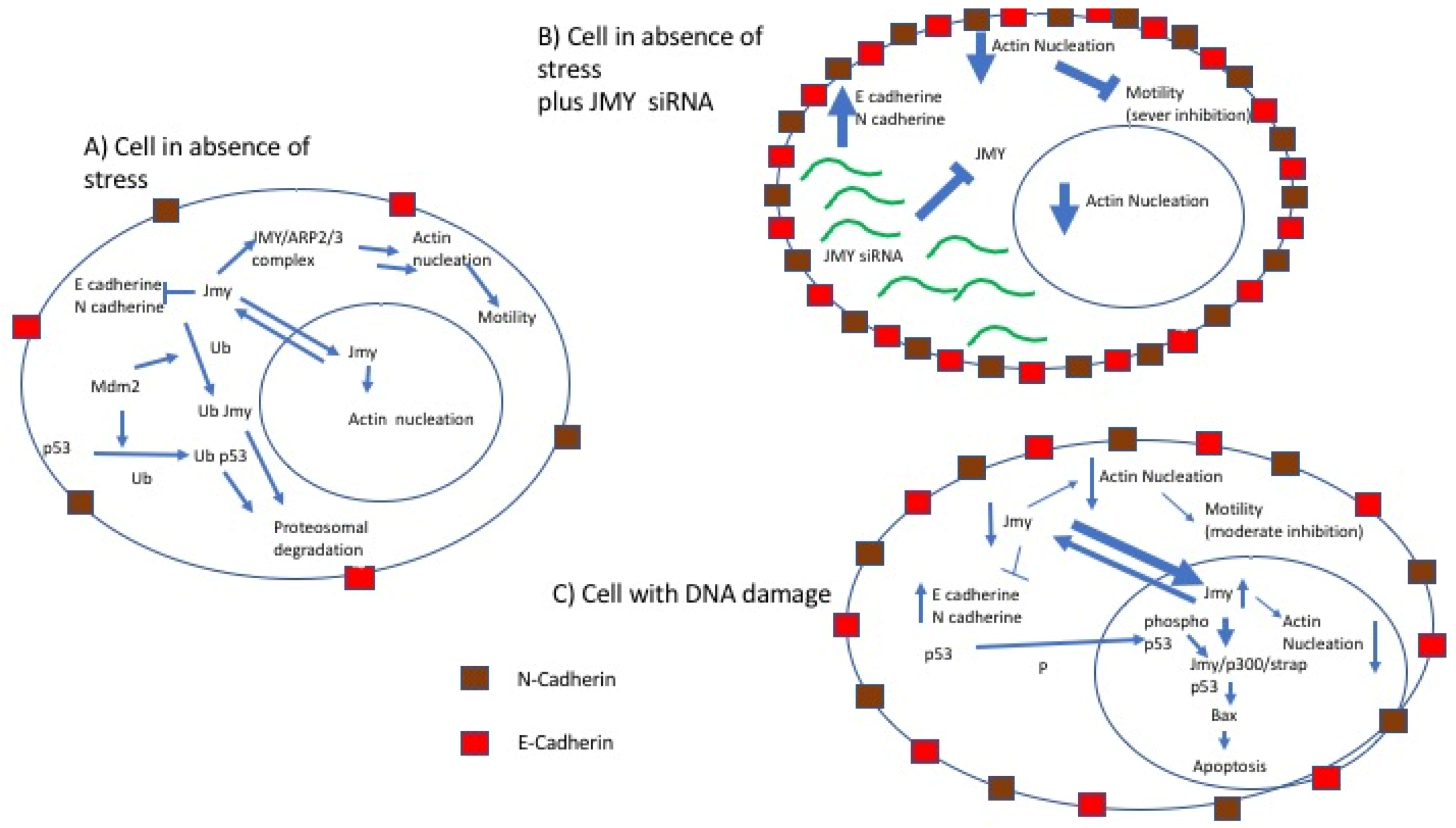
2.5. JMY and Cell Motility
3. Linking p53 Pathway and Cell Motility
4. Conclusions
Funding
Conflicts of Interest
References
- Finlay, C.A.; Hinds, P.W.; Levine, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 57, 1083–1093. [Google Scholar] [CrossRef]
- Bossi, G.; Lapi, E.; Strano, S.; Rinaldo, C.; Blandino, G.; Sacchi, A. Mutant p53 gain of function: Reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 2006, 25, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Gish, K.; Murphy, M.; Yin, Y.; Notterman, D.; Hoffman, W.H.; Tom, E.; Mack, D.H.; Levine, A.J. The transcriptional program following p53 activation. Cold Spring Harbor Symp. Quant. Biol. 2000, 65, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Coutts, A.S.; Boulahbel, H.; Graham, A.; La Thangue, N.B. MDM2 targets the p53 transcription cofactor jmy for degradation. EMBO Rep. 2007, 8, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G.; Sacchi, A. Restoration of wild-type p53 function in human cancer: Relevance for tumor therapy. Head Neck 2007, 29, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Coutts, A.S.; La Thangue, N.B. MDM2 widens its repertoire. Cell Cycle 2007, 6, 827–829. [Google Scholar] [CrossRef] [PubMed]
- KEGG (Kyoto Encyclopedia of Genes and Genoma). P53 Pathway hsa04115. Available online: http://www.kegg.jp/kegg-bin/show_pathway?map=hsa04115&show_description=show (accessed on 29 May 2018).
- Coutts, A.S.; La Thangue, N.B. The p53 response: Emerging levels of co-factor complexity. Biochem. Biophys. Res. Commun. 2005, 331, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Shikama, N.; Lee, C.W.; France, S.; Delavaine, L.; Lyon, J.; Krstic-Demonacos, M.; La Thangue, N.B. A novel cofactor for p300 that regulates the p53 response. Mol. Cell 1999, 4, 365–376. [Google Scholar] [CrossRef]
- Coutts, A.S.; Weston, L.; La Thangue, N.B. A transcription co-factor integrates cell adhesion and motility with the p53 response. Proc. Natl. Acad. Sci. USA 2009, 106, 19872–19877. [Google Scholar] [CrossRef] [PubMed]
- Adighibe, O.; Turley, H.; Leek, R.; Harris, A.; Coutts, A.S.; La Thangue, N.; Gatter, K.; Pezzella, F. JMY protein, a regulator of p53 and cytoplasmic actin filaments, is expressed in normal and neoplastic tissues. Virchows Arch. 2014, 465, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Coutts, A.S.; Quinlan, M.E.; Thangue, N.B.; Mullins, R.D. P53-cofactor JMY is a multifunctional actin nucleation factor. Nat. Cell Biol. 2009, 11, 451–459. [Google Scholar] [CrossRef] [PubMed]
- D’Souza-Schorey, C. Disassembling adherens junctions: Breaking up is hard to do. Trends Cell Biol. 2005, 15, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Jeanes, A.; Gottardi, C.J.; Yap, A.S. Cadherins and cancer: How does cadherin dysfunction promote tumor progression? Oncogene 2008, 27, 6920–6929. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hu, J.; Agorreta, J.; Cesario, A.; Zhang, Y.; Harris, A.L.; Gatter, K.; Pezzella, F. Tumor necrosis factor receptor-associated protein 1 (trap1) regulates genes involved in cell cycle and metastases. Cancer Lett. 2010, 296, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, J.; Palander, O.; Seifried, L.A.; Dick, F.A. DNA damage signals through differentially modified E2F1 molecules to induce apoptosis. Mol. Cell Biol. 2012, 32, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Hershko, T.; Chaussepied, M.; Oren, M.; Ginsberg, D. Novel link between E2F and p53: Proapoptotic cofactors of p53 are transcriptionally upregulated by e2f. Cell Death Differ. 2005, 12, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, D. E2F1 pathway to apoptosis. FEBS Lett. 2002, 529, 122–125. [Google Scholar] [CrossRef]
- Demonacos, C.; Krstic-Demonacos, M.; La Thangue, N.B. A TPR motif cofactor contributes to p300 activity in the p53 response. Mol. Cell 2001, 8, 71–84. [Google Scholar] [CrossRef]
- Demonacos, C.; Krstic-Demonacos, M.; Smith, L.; Xu, D.; O’Connor, D.P.; Jansson, M.; La Thangue, N. A new effector pathway links ATM kinase with the DNA damage response. Nat. Cell Biol. 2004, 6, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Belin, B.; Mullins, R.D. Actin binding to WH2 domains regulates nuclear import of the multifunctional actin regulator JMY. Mol. Biol. Cell 2012, 23, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. Out of the jaws of death: Prmt5 steers p53. Nat. Cell Biol. 2008, 10, 1389–1390. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.; Durant, S.T.; Cho, E.-C.; Sheahan, S.; Edelmann, M.; Kessler, B.; La Thangue, N.B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.C.; Zheng, S.; Munro, S.; Liu, G.; Carr, S.M.; Moehlenbrink, J.; Lu, Y.C.; Stimson, L.; Khan, O.; Konietzny, R.; et al. Arginine methylation controls growth regulation by e2f-1. EMBO J. 2012, 31, 1785–1797. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 c-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Roadcap, D.W.; Bear, J.E. Double JMY: Making actin fast. Nat. Cell Biol. 2009, 11, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Coutts, A.S.; Pires, I.M.; Weston, L.; Buffa, F.M.; Milani, M.; Li, J.L.; Harris, A.L.; Hammond, E.M.; La Thangue, N.B. Hypoxia-driven cell motility reflects the interplay between JMY and hif-1alpha. Oncogene 2011, 30, 4835–4842. [Google Scholar] [CrossRef] [PubMed]
- Firat-Karalar, E.N.; Hsiue, P.P.; Welch, M.D. The actin nucleation factor JMY is a negative regulator of neuritogenesis. Mol. Biol. Cell 2011, 22, 4563–4574. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A. Jasplakinolide: An actin-specific reagent that promotes actin polymerization. Methods Mol. Biol. 2009, 586, 71–87. [Google Scholar] [PubMed]
- Ishimoto, T.; Ozawa, T.; Mori, H. Real-time monitoring of actin polymerization in living cells using split luciferase. Bioconjug. Chem. 2011, 22, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.; Gadea, G.; Roux, P. Control of cell migration: A tumour suppressor function for p53? Biol. Cell 2006, 98, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Jimmy on the stage: Linking DNA damage with cell adhesion and motility. Cell Adh. Migr. 2010, 4, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Coutts, A.S.; Weston, L.; La Thangue, N.B. Actin nucleation by a transcription co-factor that links cytoskeletal events with the p53 response. Cell Cycle 2010, 9, 1511–1515. [Google Scholar] [CrossRef] [PubMed]
- Nurnberg, A.; Kitzing, T.; Grosse, R. Nucleating actin for invasion. Nat. Rev. Cancer 2011, 11, 177–187. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adighibe, O.; Pezzella, F. The Role of JMY in p53 Regulation. Cancers 2018, 10, 173. https://doi.org/10.3390/cancers10060173
Adighibe O, Pezzella F. The Role of JMY in p53 Regulation. Cancers. 2018; 10(6):173. https://doi.org/10.3390/cancers10060173
Chicago/Turabian StyleAdighibe, Omanma, and Francesco Pezzella. 2018. "The Role of JMY in p53 Regulation" Cancers 10, no. 6: 173. https://doi.org/10.3390/cancers10060173
APA StyleAdighibe, O., & Pezzella, F. (2018). The Role of JMY in p53 Regulation. Cancers, 10(6), 173. https://doi.org/10.3390/cancers10060173