Rac-Mediated Macropinocytosis of Extracellular Protein Promotes Glucose Independence in Non-Small Cell Lung Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
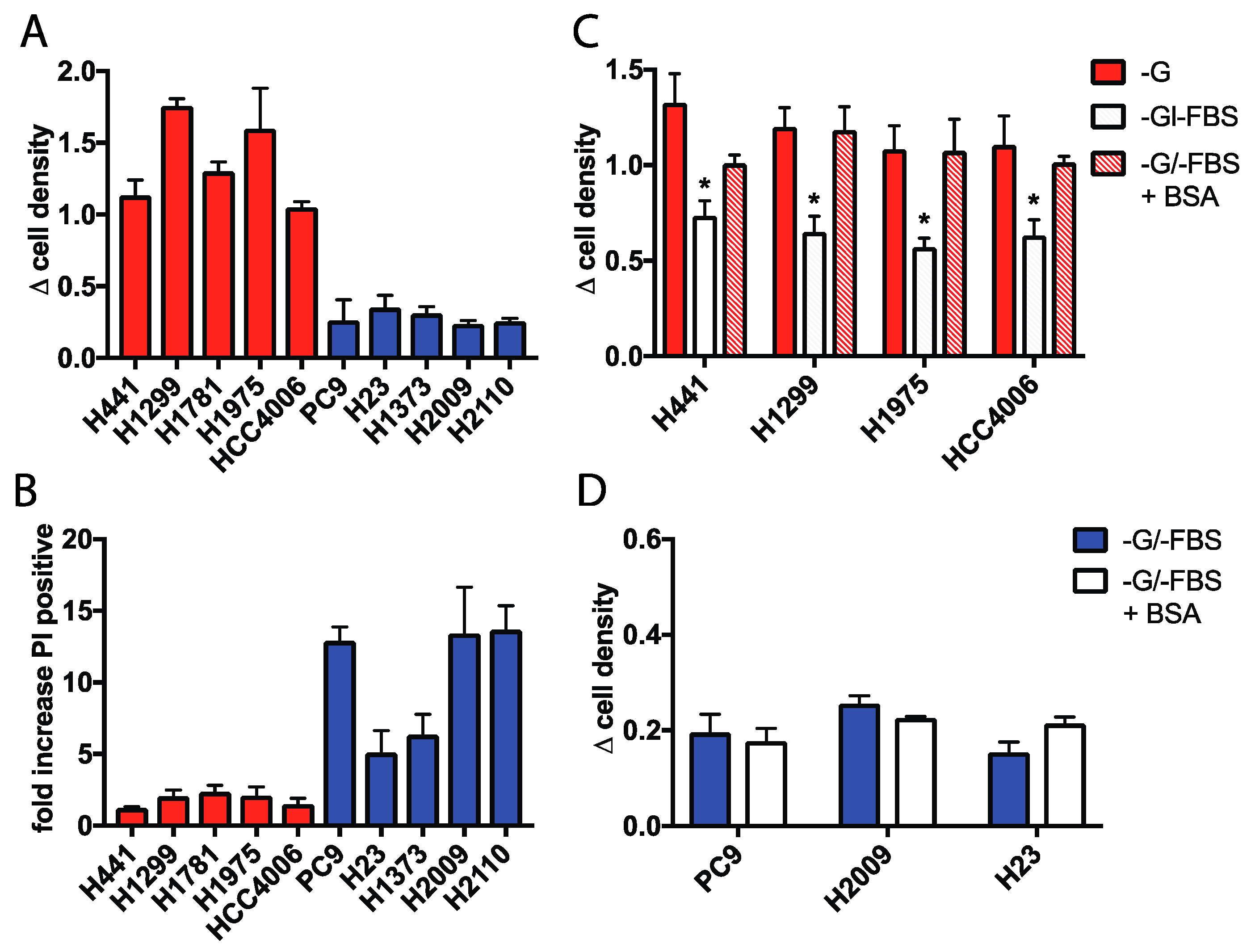
2.1. Glucose-Independent NSCLC Cells Require Extracellular Protein for Growth During Glucose Withdrawal
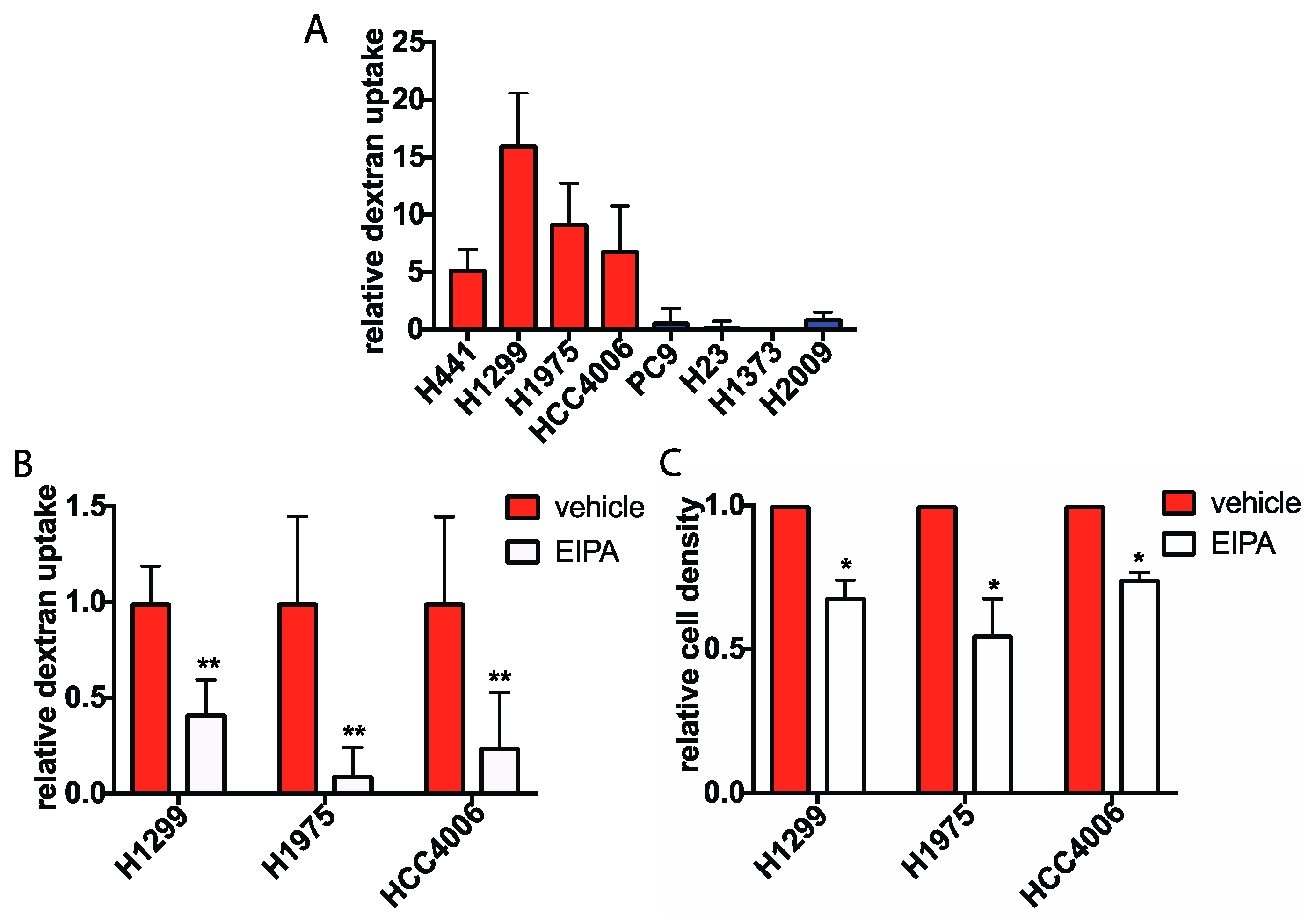
2.2. Macropinocytosis Is Increased in Glucose Independent Cells and Is Required for Growth in the Absence of Glucose
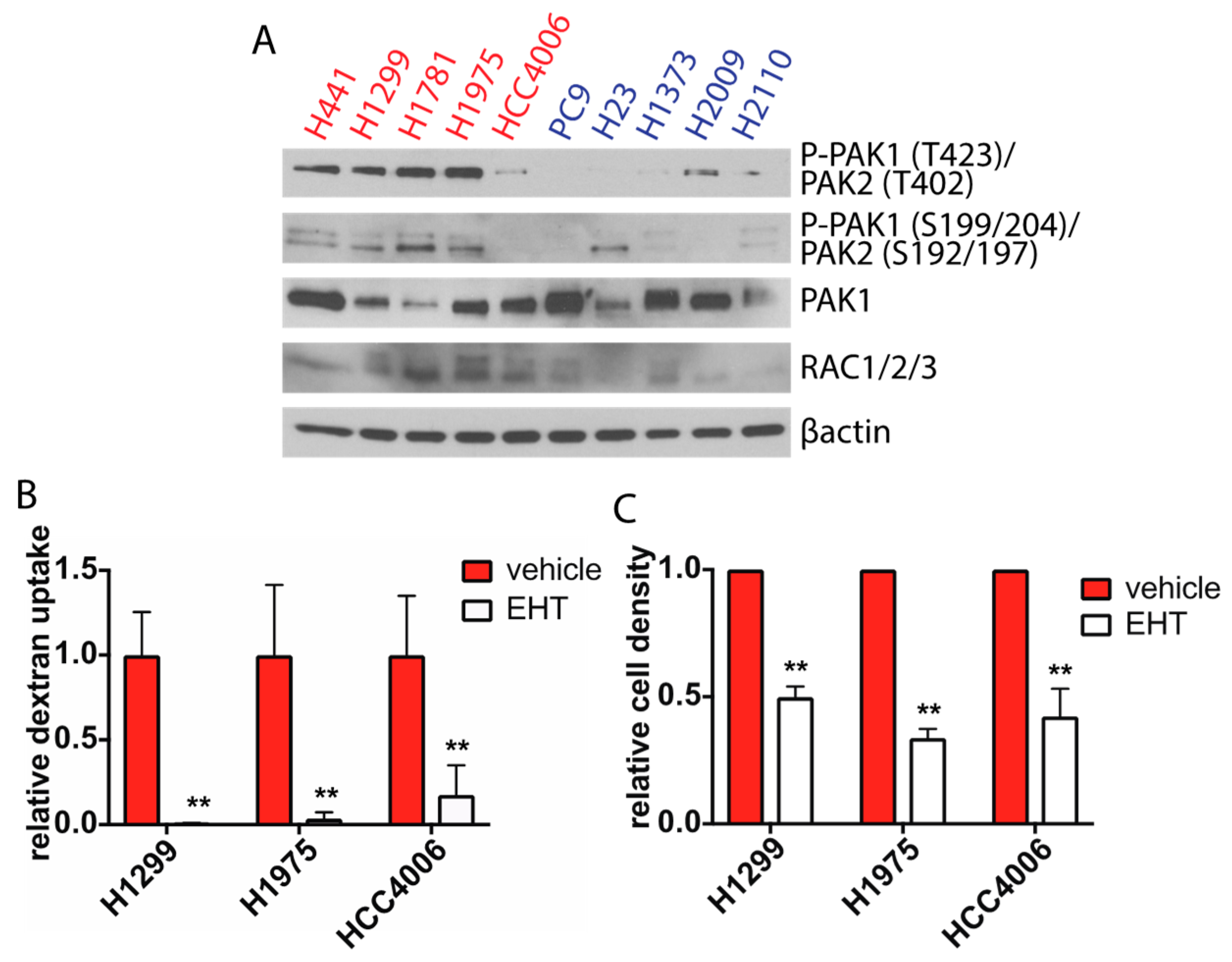
2.3. Rac Signaling is Elevated in Glucose Independent Cells and Inhibition of Rac Decreases Macropinocytosis and Glucose-Free Survival
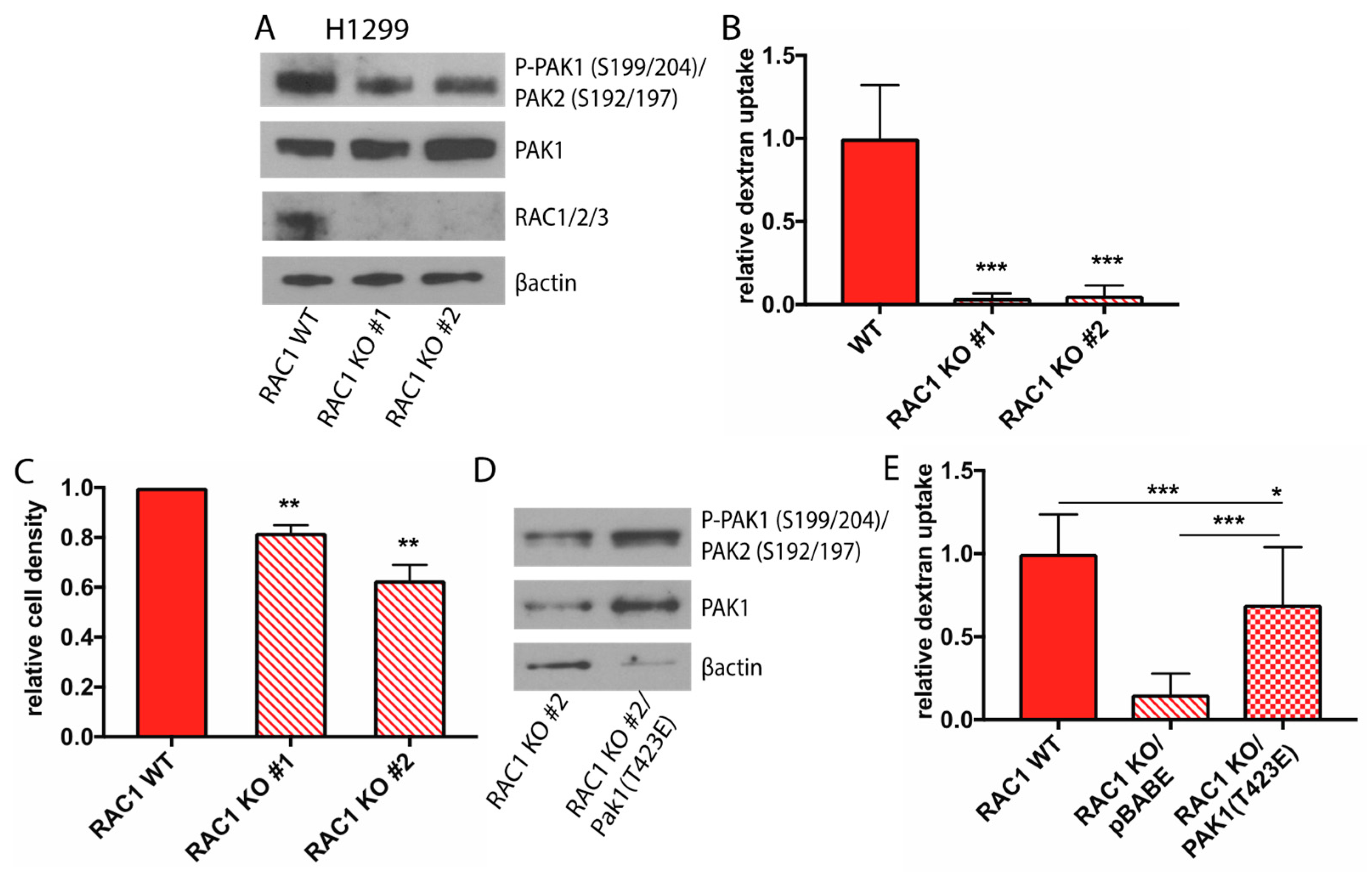
2.4. Rac1 Expression Regulates Macropinocytosis in a Pak Dependent Manner and is Necessary for Glucose Independence
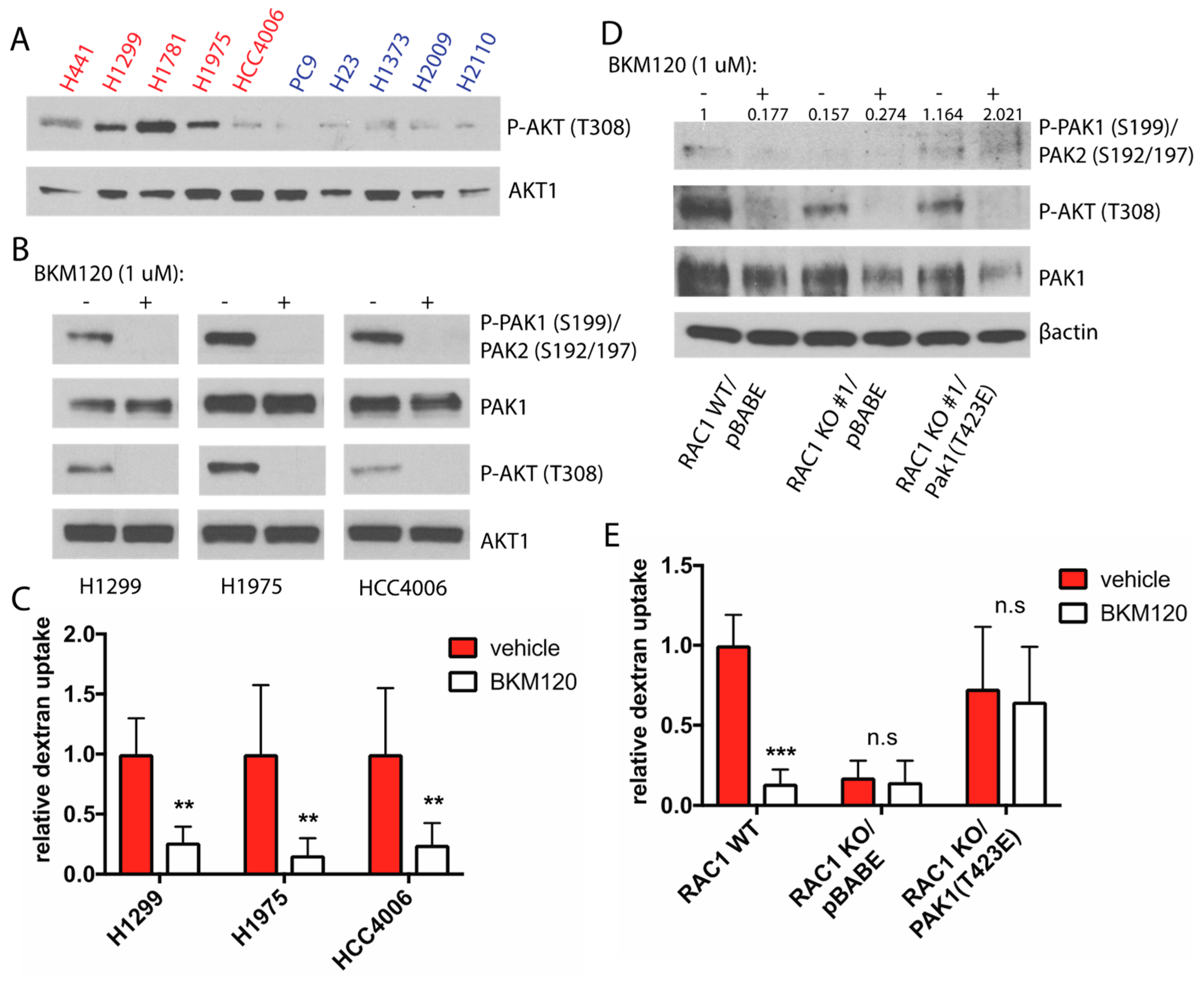
2.5. PI3K Regulates Rac-Driven Macropinocytosis
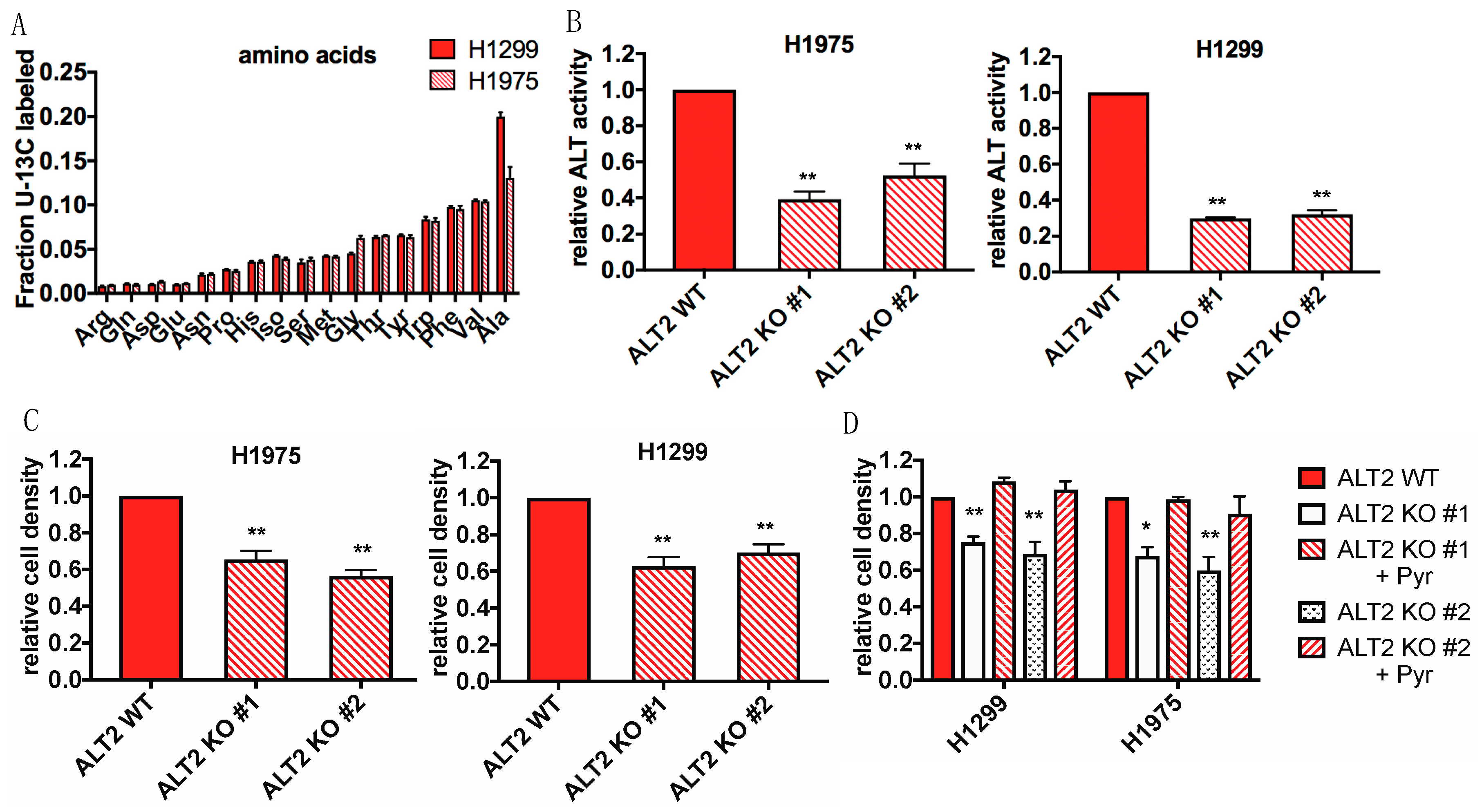
2.6. Internalized Protein Supplies Glucose Starved Cells with Alanine and Other Free Amino Acids
2.7. ALT2 Activity is Critical for Proliferation During Glucose Starvation
3. Discussion
4. Materials and Methods
4.1. Reagents and Plasmids
4.2. Cell Culture
4.3. Cell Line Generation
4.4. Generation of Retroviral PAK (T423E) Stable Cell Lines
4.5. Xenograft Assays
4.6. Immunoblotting
4.7. Glucose-Free Growth Assays
4.8. Macropinocytosis Visualization and Quantification
4.9. Generation of 13C-Labeled Proteins
4.10. Metabolic Tracing and Extraction
4.11. Liquid Chromatography/Mass Spectrometry (LC/MS) Analysis
4.12. ALT Activity Assay
4.13. Public Dataset: Cancer Cell Line Encyclopedia (CCLE) Data Analysis
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Ann. Rev. Cell. Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gullino, P.M.; Grantham, F.H.; Courtney, A.H. Glucose Consumption by Transplanted Tumors in Vivo. Cancer Res. 1967, 27, 1031–1040. [Google Scholar] [PubMed]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Urasaki, Y.; Heath, L.; Xu, C.W. Coupling of Glucose Deprivation with Impaired Histone H2B Monoubiquitination in Tumors. PLoS ONE 2012, 7, e36775. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.M.; Barros, A.S.; Gil, A.M.; Goodfellow, B.J.; Humpfer, E.; Spraul, M.; Carreira, I.M.; Melo, J.B.; Bernardo, J.; Gomes, A.; et al. Metabolic Profiling of Human Lung Cancer Tissue by 1H High Resolution Magic Angle Spinning (HRMAS) NMR Spectroscopy. J. Proteome Res. 2010, 9, 319–332. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Cantley, L.C. Cancer’s Fuel Choice: New Flavors for a Picky Eater. Mol. Cell. 2015, 60, 514–523. [Google Scholar] [CrossRef]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.F.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Target Validation Team. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015, 35, S129–S150. [Google Scholar] [CrossRef]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine Oxidation Maintains the TCA Cycle and Cell Survival during Impaired Mitochondrial Pyruvate Transport. Mol. Cell. 2014, 56, 414–424. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Pelicano, H.; Huang, P. Cancer Metabolism: Is Glutamine Sweeter than Glucose? Cancer Cell. 2010, 18, 199–200. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montal, E.D.; Dewi, R.; Bhalla, K.; Ou, L.; Hwang, B.J.; Ropell, A.E.; Gordon, C.; Liu, W.J.; DeBerardinis, R.J.; Sudderth, J.; et al. PEPCK Coordinates the Regulation of Central Carbon Metabolism to Promote Cancer Cell Growth. Mol. Cell. 2015, 60, 571–583. [Google Scholar] [CrossRef]
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.C.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choinière, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell. 2015, 60, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leithner, K.; Hrzenjak, A.; Trötzmüller, M.; Moustafa, T.; Köfeler, H.C.; Wohlkoenig, C.; Stacher, E.; Lindenmann, J.; Harris, A.L.; Olschewski, A.; et al. PCK2 activation mediates an adaptive response to glucose depletion in lung cancer. Oncogene 2014, 34, 1044–1050. [Google Scholar] [CrossRef]
- Balsa-Martinez, E.; Puigserver, P. Cancer Cells Hijack Gluconeogenic Enzymes to Fuel Cell Growth. Mol. Cell. 2015, 60, 509–511. [Google Scholar] [CrossRef]
- Chen, J.; Lee, H.J.; Wu, X.; Huo, L.; Kim, S.J.; Xu, L.; Wang, Y.; He, J.; Bollu, L.R.; Gao, G.; et al. Gain of Glucose-Independent Growth upon Metastasis of Breast Cancer Cells to the Brain. Cancer Res. 2015, 75, 554–565. [Google Scholar] [CrossRef]
- Li, B.; Qiu, B.; Lee, D.S.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Vincent, E.E.; Griss, T.; Samborska, B.; Izreig, S.; Svensson, R.U.; Mamer, O.A.; Avizonis, D.; Shackelford, D.B.; Shaw, R.J.; et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc. Natl. Acad. Sci. USA 2014, 111, 2554–2559. [Google Scholar] [CrossRef]
- Fleck, A.; Hawker, F.; Wallace, P.I.; Raines, G.; Trotter, J.; Ledingham, I.M.; Calman, K.C. Increased vascular permeability: A major cause of hypoalbuminaemia in disease and injury. Lancet 1985, 325, 781–784. [Google Scholar] [CrossRef]
- Stehle, G.; Sinn, H.; Wunder, A.; Schrenk, H.H.; Stewart, J.C.; Hartung, G.; Maier-Borst, W.; Heene, D.L. Plasma protein (albumin) catabolism by the tumor itself—Implications for tumor metabolism and the genesis of cachexia. Crit. Rev. Oncol. Hematol. 1997, 26, 77–100. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Feramisco, J. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 1986, 233, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; Del Rosario, A.M.; Whitman, M.; Chin, C.R.; et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nature 2016, 23, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Racoosin, E.L. Macropinosome maturation and fusion with tubular lysosomes in macrophages. J. Cell. Biol. 1993, 121, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Koivusalo, M.; Welch, C.; Hayashi, H.; Scott, C.C.; Kim, M.; Alexander, T.; Touret, N.; Hahn, K.M.; Grinstein, S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell. Biol. 2010, 188, 547–563. [Google Scholar] [CrossRef] [Green Version]
- Canton, J.; Schlam, D.; Breuer, C.; Gütschow, M.; Glogauer, M.; Grinstein, S. Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Dharmawardhane, S.; Schürmann, A.; Sells, M.A.; Chernoff, J.; Schmid, S.L.; Bokoch, G.M. Regulation of macropinocytosis by p21-activated kinase-1. Mol. Biol. Cell. 2000, 11, 3341–3352. [Google Scholar] [CrossRef] [PubMed]
- West, M.A.; Prescott, A.R.; Eskelinen, E.L.; Ridley, A.J.; Watts, C. Rac is required for constitutive macropinocytosis by dendritic cells but does not control its downregulation. Curr. Biol. 2000, 10, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Palm, W.; Araki, J.; King, B.; DeMatteo, R.G.; Thompson, C.B. Critical role for PI3-kinase in regulating the use of proteins as an amino acid source. Proc. Natl. Acad. Sci. USA 2017, 114, E8628–E8636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eby, J.J.; Holly, S.P.; Van Drogen, F.; Grishin, A.V.; Peter, M.; Drubin, D.G.; Blumer, K.J. Actin cytoskeleton organization regulated by the PAK family of protein kinases. Curr. Biol. 1998, 8, 967–970. [Google Scholar] [CrossRef]
- Sells, M.A.; Knaus, U.G.; Bagrodia, S.; Ambrose, D.M.; Bokoch, G.M.; Chernoff, J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 1997, 7, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Manser, E.; Huang, H.Y.; Loo, T.H.; Chen, X.Q.; Dong, J.M.; Leung, T.; Lim, L. Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol. Cell. Biol. 1997, 17, 1129–1143. [Google Scholar] [CrossRef]
- Knaus, U.G.; Wang, Y.; Reilly, A.M.; Warnock, D.; Jackson, J.H. Structural Requirements for PAK Activation by Rac GTPases. J. Biol. Chem. 1998, 273, 21512–21518. [Google Scholar] [CrossRef] [Green Version]
- Knaus, U.G.; Bokoch, G.M. The p21Rac/Cdc42-activated kinases (PAKs). Int. J. Biochem. Cell. Biol. 1998, 30, 857–862. [Google Scholar] [CrossRef]
- Onesto, C.; Shutes, A.; Picard, V.; Schweighoffer, F.; Der, C.J. Characterization of EHT 1864, a Novel Small Molecule Inhibitor of Rac Family Small GTPases. Methods Enzymol. 2008, 439, 111–129. [Google Scholar] [CrossRef]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and Mechanism of Action of EHT 1864, a Novel Small Molecule Inhibitor of Rac Family Small GTPases. J. Biol. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef] [Green Version]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac pathways. Small GTPases 2015, 6, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, K.; Zhong, S.; Xie, K.; Yu, D.; Yang, R.; Gong, D.W. Hepatic ALT isoenzymes are elevated in gluconeogenic conditions including diabetes and suppressed by insulin at the protein level. Diabetes Metab. Res. Rev. 2015, 31, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Lin, S.H.; Wang, Y.; Chin, Y.E.; Kang, L.; Mi, J. Glutamic Pyruvate Transaminase GPT2 Promotes Tumorigenesis of Breast Cancer Cells by Activating Sonic Hedgehog Signaling. Theranostics 2017, 7, 3021–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coloff, J.L.; Murphy, J.P.; Braun, C.R.; Harris, I.S.; Shelton, L.M.; Kami, K.; Gygi, S.P.; Selfors, L.M.; Brugge, J.S. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell. Metab. 2016, 23, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.; Schafer, X.L.; Ambeskovic, A.; Spencer, C.M.; Land, H.; Munger, J. Addiction to Coupling of the Warburg Effect with Glutamine Catabolism in Cancer Cells. Cell. Rep. 2016, 17, 821–836. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Wang, X.; Liu, Y.; Li, Y.; Colvin, R.A.; Tong, L.; Wu, S.; Chen, X. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett. 2014, 351, 242–251. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2015, 150, 251–263. [Google Scholar] [CrossRef]
- Alan, J.K.; Lundquist, E.A. Mutationally activated Rho GTPases in cancer. Small GTPases 2014, 4, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2014, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Brachetti, C.; Bahlmann, F.; Schmidt, M.; Kaina, B. Rho GTPases in human breast tumours: Expression and mutation analyses and correlation with clinical parameters. Brit. J. Cancer 2002, 87, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.L.; Hong, Y.R.; Sy, W.D.; Lieu, A.S.; Lin, C.L.; Lee, K.S.; Howng, S.L. Rac1 gene mutations in human brain tumours. EJSO 2004, 30, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Bin Shieh, D.; Godleski, J.; Herndon, J.E., 2nd; Azuma, T.; Mercer, H.; Sugarbaker, D.J.; Kwiatkowski, D.J. Cell motility as a prognostic factor in Stage I nonsmall cell lung carcinoma. Cancer 1999, 85, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Bi, F.; Liu, N.; Xue, Y.; Yao, X.; Zheng, Y.; Fan, D. Expression of seven main Rho family members in gastric carcinoma. Biochem. Biophys. Res. Commun. 2004, 315, 686–691. [Google Scholar] [CrossRef]
- Kamai, T. Overexpression of RhoA, Rac1, and Cdc42 GTPases Is Associated with Progression in Testicular Cancer. Clin. Cancer Res. 2004, 10, 4799–4805. [Google Scholar] [CrossRef] [Green Version]
- Fritz, G.; Just, I.; Kaina, B. Rho GTPases are over-expressed in human tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef] [Green Version]
- Oxender, D.L.; Christensen, H.N. Evidence for two Types of Mediation of Neutral Amino-Acid Transport in Ehrlich Cells. Nature 1963, 197, 765–767. [Google Scholar] [CrossRef]
- Barker, G.A.; Ellory, J.C. The identification of neutral amino acid transport systems. Exp. Physiol. 1990, 75, 3–26. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Commisso, C.; Flinn, R.J.; Bar-Sagi, D. Determining the macropinocytic index of cells through a quantitative image-based assay. Nat. Protoc. 2014, 9, 182–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, M.D.; Hwang, S.K.; Pauli, C.; Murphy, C.J.; Cheng, Z.; Hopkins, B.D.; Wu, D.; Loughran, R.M.; Emerling, B.M.; Zhang, G.; et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E743–E752. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hodakoski, C.; Hopkins, B.D.; Zhang, G.; Su, T.; Cheng, Z.; Morris, R.; Rhee, K.Y.; Goncalves, M.D.; Cantley, L.C. Rac-Mediated Macropinocytosis of Extracellular Protein Promotes Glucose Independence in Non-Small Cell Lung Cancer. Cancers 2019, 11, 37. https://doi.org/10.3390/cancers11010037
Hodakoski C, Hopkins BD, Zhang G, Su T, Cheng Z, Morris R, Rhee KY, Goncalves MD, Cantley LC. Rac-Mediated Macropinocytosis of Extracellular Protein Promotes Glucose Independence in Non-Small Cell Lung Cancer. Cancers. 2019; 11(1):37. https://doi.org/10.3390/cancers11010037
Chicago/Turabian StyleHodakoski, Cindy, Benjamin D. Hopkins, Guoan Zhang, Taojunfeng Su, Zhe Cheng, Roxanne Morris, Kyu Y. Rhee, Marcus D. Goncalves, and Lewis C. Cantley. 2019. "Rac-Mediated Macropinocytosis of Extracellular Protein Promotes Glucose Independence in Non-Small Cell Lung Cancer" Cancers 11, no. 1: 37. https://doi.org/10.3390/cancers11010037
APA StyleHodakoski, C., Hopkins, B. D., Zhang, G., Su, T., Cheng, Z., Morris, R., Rhee, K. Y., Goncalves, M. D., & Cantley, L. C. (2019). Rac-Mediated Macropinocytosis of Extracellular Protein Promotes Glucose Independence in Non-Small Cell Lung Cancer. Cancers, 11(1), 37. https://doi.org/10.3390/cancers11010037