Constitutive BRCA1 Promoter Hypermethylation Can Be a Predisposing Event in Isolated Early-Onset Breast Cancer

 , , , , , ,
, , , , , ,  , ,
, ,
Abstract
:1. Introduction
2. Results
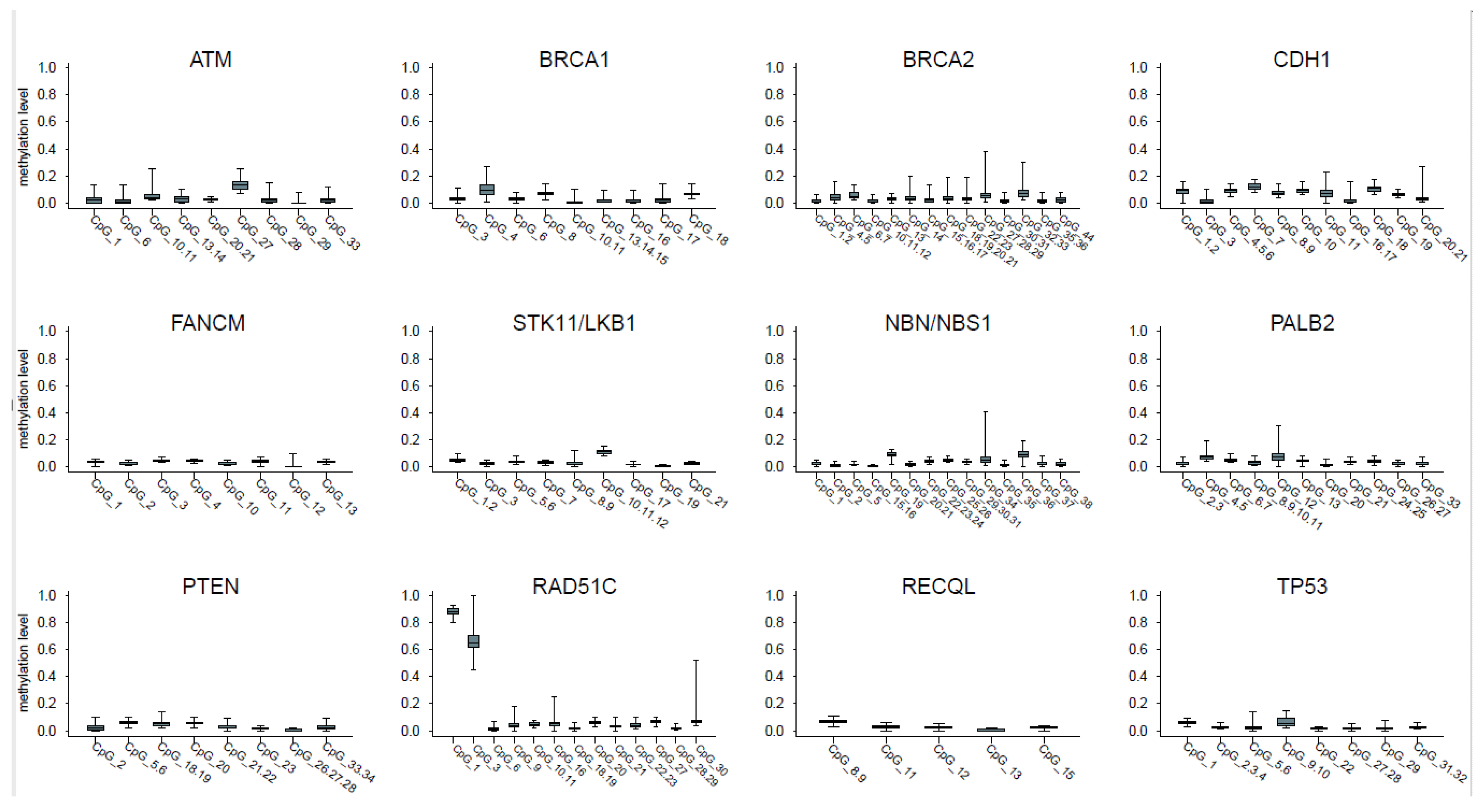
2.1. CpGs Selection
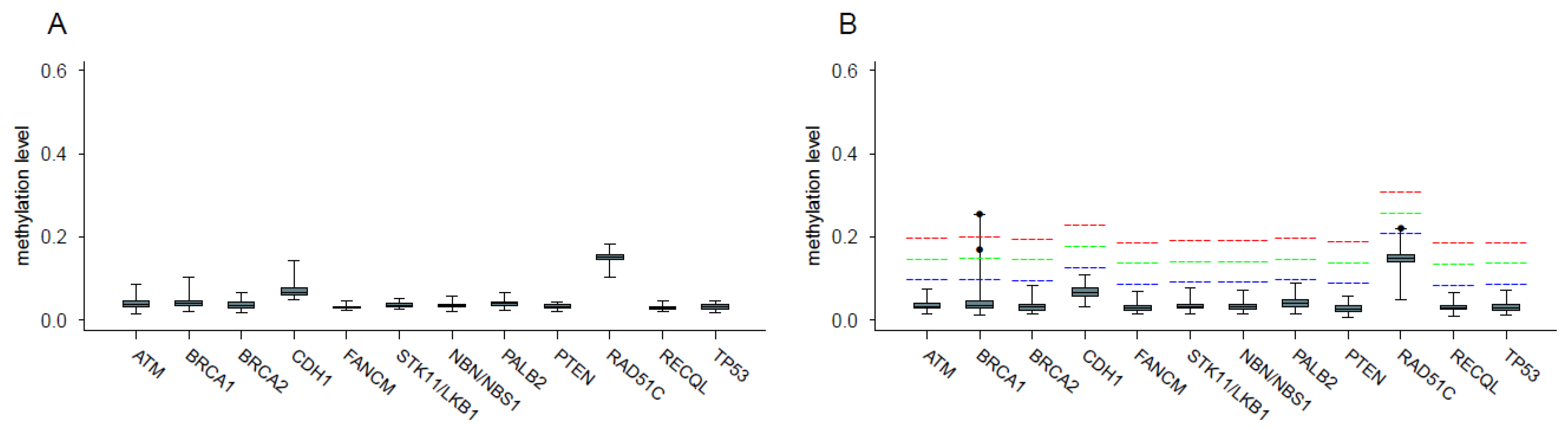
2.2. Analysis of Promoter Methylation in Control Samples
2.3. Analysis of Promoter Methylation in Patients and Identification of Hypermethylated Cases
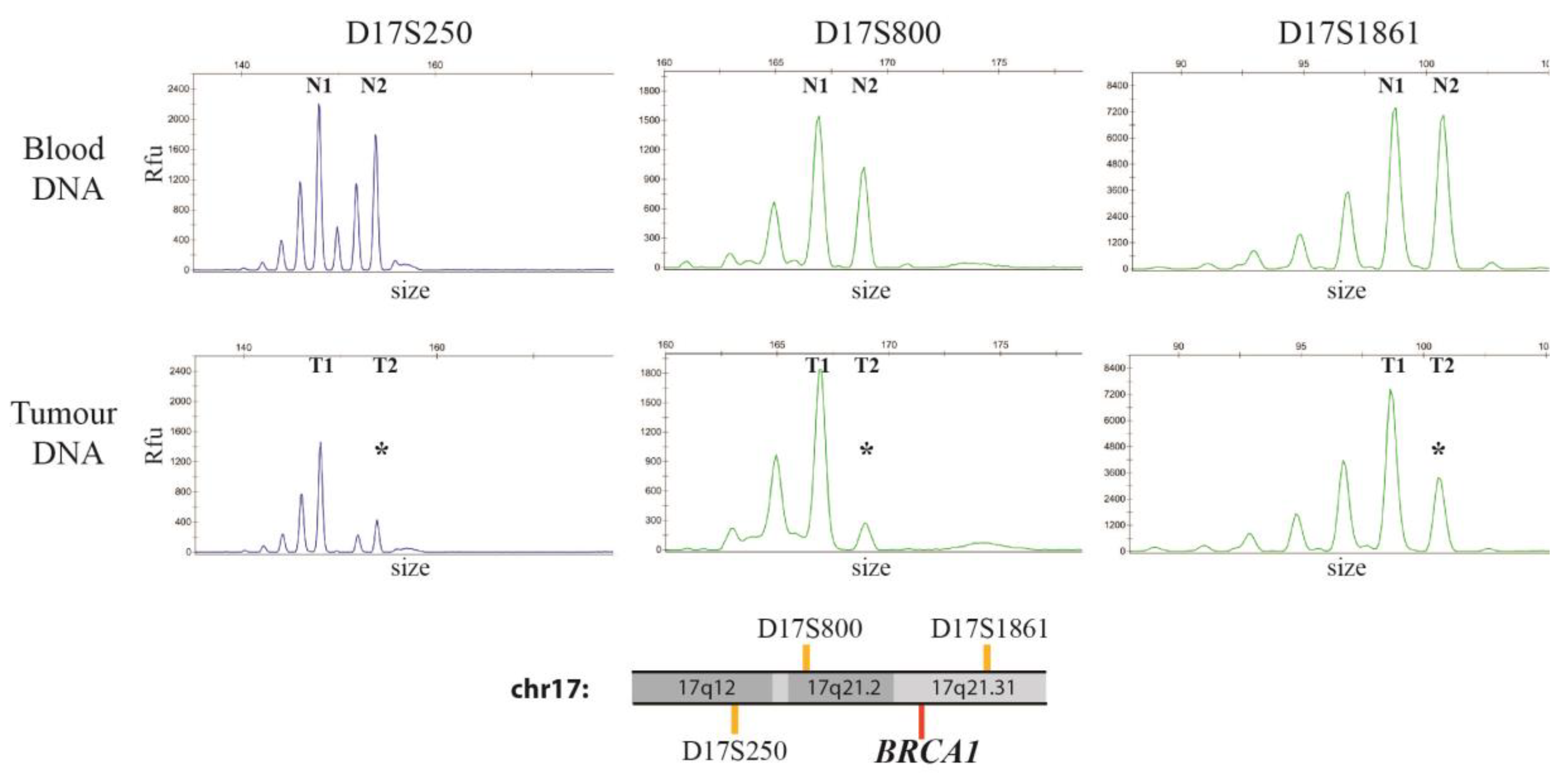
2.4. Assessment of Loss of Heterozygosity (LOH)
2.5. Relationship between Methylation Levels, Age at Blood Withdrawal and Chemotherapy
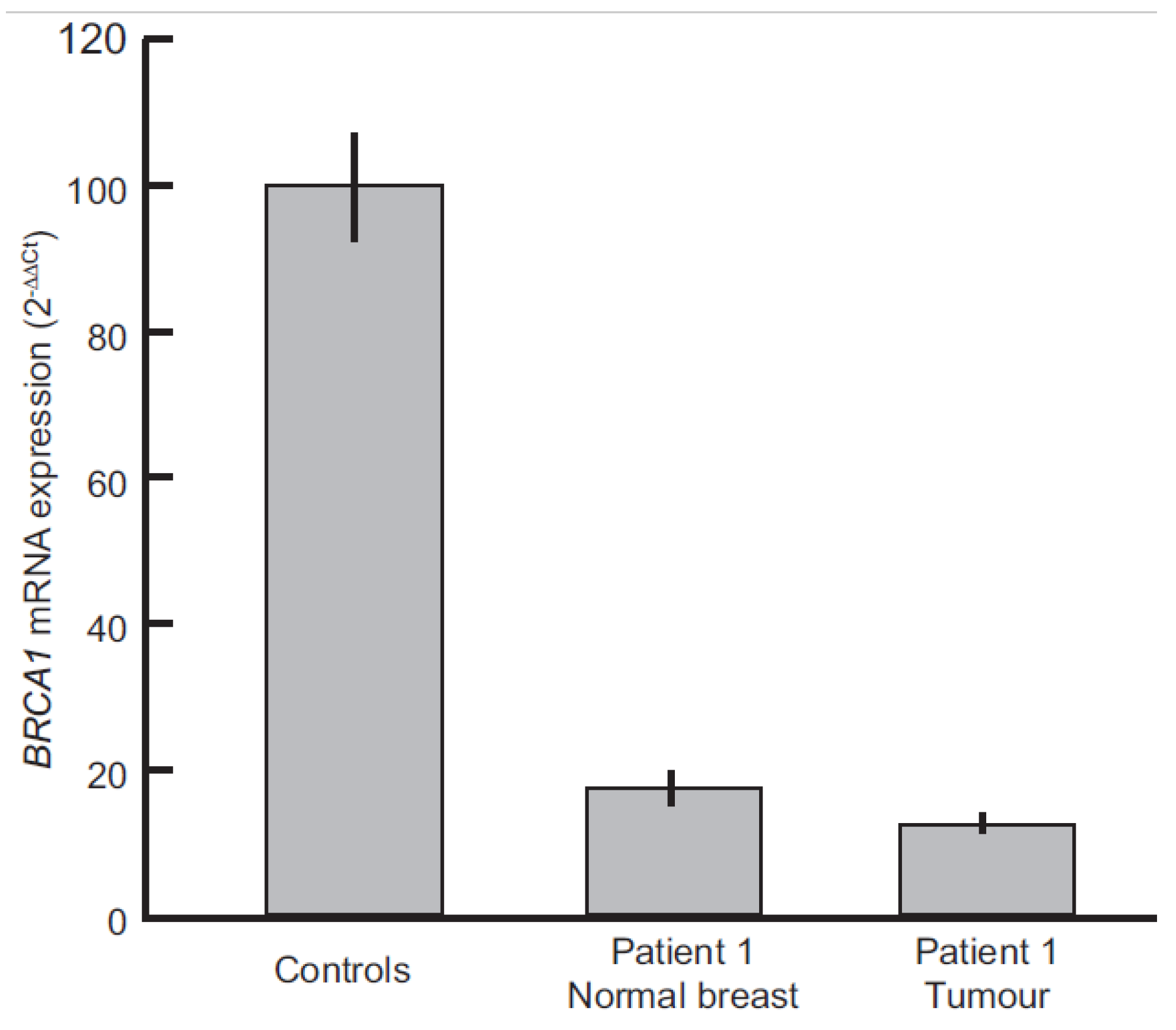
2.6. Evaluation of BRCA1 Expression Levels
3. Discussion
4. Methods
4.1. Study Subjects
4.2. DNA Extraction and Bisulfite Conversion
4.3. Primer Design and Mass Spectrometry
4.4. CpG Selection
4.5. Statistical Methods
4.6. Evaluation of LOH by STRs Analysis
4.7. BRCA1 Promoter Sequencing
4.8. mRNA Extraction and BRCA1 Expression Quantification by Real-Time PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources; methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Fidler, M.M.; Gupta, S.; Soerjomataram, I.; Ferlay, J.; Steliarova-Foucher, E.; Bray, F. Cancer incidence and mortality among young adults aged 20–39 years worldwide in 2012: A population-based study. Lancet Oncol. 2017, 18, 1579–1589. [Google Scholar] [CrossRef]
- AIRTUM (Associazione Italiana Registro Tumori) Cancer Registry—2017. Available online: http://www.registri-tumori.it/PDF/AIOM2017/2017_numeri_del_cancro.pdf (accessed on 21 August 2018).
- Kleibl, Z.; Kristensen, V.N. Women at high risk of breast cancer: Molecular characteristics; clinical presentation and management. Breast 2016, 28, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Möller, S.; Unger, R.H.; et al. Nordic Twin Study of Cancer (NorTwinCan) Collaboration. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef]
- Meindl, A. German Consortium for Hereditary Breast and Ovarian Cancer. Comprehensive analysis of 989 patients with breast or ovarian cancer provides BRCA1 and BRCA2 mutation profiles and frequencies for the German population. Int. J. Cancer 2002, 97, 472–480. [Google Scholar] [PubMed]
- Han, S.A.; Park, S.K.; Ahn, S.H.; Lee, M.H.; Noh, D.Y.; Kim, L.S.; Noh, W.C.; Jung, Y.; Kim, K.S.; Kim, S.W.; et al. The Korean Hereditary Breast Cancer (KOHBRA) study: Protocols and interim report. Clin. Oncol. (R. Coll. Radiol.) 2011, 23, 434–441. [Google Scholar] [CrossRef] [PubMed]
- De Leeneer, K.; Coene, I.; Crombez, B.; Simkens, J.; Van den Broecke, R.; Bols, A.; Stragier, B.; Vanhoutte, I.; De Paepe, A.; Poppe, B.; et al. Prevalence of BRCA1/2 mutations in sporadic breast/ovarian cancer patients and identification of a novel de novo BRCA1 mutation in a patient diagnosed with late onset breast and ovarian cancer: Implications for genetic testing. Breast Cancer Res. Treat. 2012, 132, 87–95. [Google Scholar] [CrossRef]
- Varesco, L.; Viassolo, V.; Viel, A.; Gismondi, V.; Radice, P.; Montagna, M.; Alducci, E.; Della Puppa, L.; Oliani, C.; Tommasi, S.; et al. Performance of BOADICEA and BRCAPRO genetic models and of empirical criteria based on cancer family history for predicting BRCA mutation carrier probabilities: A retrospective study in a sample of Italian cancer genetics clinics. Breast 2013, 22, 1130–1135. [Google Scholar] [CrossRef]
- Azzollini, J.; Scuvera, G.; Bruno, E.; Pasanisi, P.; Zaffaroni, D.; Calvello, M.; Pasini, B.; Ripamonti, C.B.; Colombo, M.; Pensotti, V.; et al. Mutation detection rates associated with specific selection criteria for BRCA1/2 testing in 1854 high-risk families: A monocentric Italian study. Eur. J. Intern. Med. 2016, 32, 65–71. [Google Scholar] [CrossRef]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients with Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Hauke, J.; Horvath, J.; Groß, E.; Gehrig, A.; Honisch, E.; Hackmann, K.; Schmidt, G.; Arnold, N.; Faust, U.; Sutter, C.; et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018, 7, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, R.L.; Kuchenbaecker, K.B.; Michailidou, K.; Beesley, J.; Kar, S.; Lindström, S.; Hui, S.; Lemaçon, A.; Soucy, P.; Dennis, J.; et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat. Genet. 2017, 49, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Miozzo, M.; Vaira, V.; Sirchia, S.M. Epigenetic alterations in cancer and personalized cancer treatment. Future Oncol. 2015, 11, 333–348. [Google Scholar] [CrossRef]
- Hitchins, M.P.; Wong, J.J.; Suthers, G.; Suter, C.M.; Martin, D.I.; Hawkins, N.J.; Ward, R.L. Inheritance of a cancer-associated MLH1 germ-line epimutation. N. Engl. J. Med. 2007, 356, 697–705. [Google Scholar] [CrossRef]
- Snell, C.; Krypuy, M.; Wong, E.M.; kConFab investigators; Loughrey, M.B.; Dobrovic, A. BRCA1 promoter methylation in peripheral blood DNA of mutation negative familial breast cancer patients with a BRCA1 tumour phenotype. Breast Cancer Res. 2008, 10, R12. [Google Scholar] [CrossRef]
- Dobrovic, A.; Kristensen, L.S. DNA methylation; epimutations and cancer predisposition. Int. J. Biochem. Cell Biol. 2009, 41, 34–39. [Google Scholar] [CrossRef]
- Galetzka, D.; Hansmann, T.; El Hajj, N.; Weis, E.; Irmscher, B.; Ludwig, M.; Schneider-Rätzke, B.; Kohlschmidt, N.; Beyer, V.; Bartsch, O.; et al. Monozygotic twins discordant for constitutive BRCA1 promoter methylation; childhood cancer and secondary cancer. Epigenetics 2012, 7, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, T.; Pliushch, G.; Leubner, M.; Kroll, P.; Endt, D.; Gehrig, A.; Preisler-Adams, S.; Wieacker, P.; Haaf, T. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum. Mol. Genet. 2012, 21, 4669–4679. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.M.; Munoz-Alegre, M.; Henderson, S.; Tang, T.; Sun, P.; Johnson, N.; Fletcher, O.; Dos Santos Silva, I.; Peto, J.; Boshoff, C.; et al. Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Hum. Mol. Genet. 2009, 18, 1332–1342. [Google Scholar] [CrossRef]
- Potapova, A.; Hoffman, A.M.; Godwin, A.K.; Al-Saleem, T.; Cairns, P. Promoter hypermethylation of the PALB2 susceptibility gene in inherited and sporadic breast and ovarian cancer. Cancer Res. 2008, 68, 998–1002. [Google Scholar] [CrossRef]
- Wong, E.M.; Southey, M.C.; Fox, S.B.; Brown, M.A.; Dowty, J.G.; Jenkins, M.A.; Giles, G.G.; Hopper, J.L.; Dobrovic, A. Constitutional methylation of the BRCA1 promoter is specifically associated with BRCA1 mutation-associated pathology in early-onset breast cancer. Cancer Prev. Res. 2011, 4, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Al-Moghrabi, N.; Nofel, A.; Al-Yousef, N.; Madkhali, S.; Amer, S.M.; Alaiya, A.; Shinwari, Z.; Al-Tweigeri, T.; Karakas, B.; Tulbah, A.; et al. The molecular significance of methylated BRCA1 promoter in white blood cells of cancer-free females. BMC Cancer 2014, 14, 830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Long, X. Association of BRCA1 promoter methylation with sporadic breast cancers: Evidence from 40 studies. Sci. Rep. 2015, 5, 17869. [Google Scholar] [CrossRef]
- Sturgeon, S.R.; Balasubramanian, R.; Schairer, C.; Muss, H.B.; Ziegler, R.G.; Arcaro, K.F. Detection of promoter methylation of tumor suppressor genes in serum DNA of breast cancer cases and benign breast disease controls. Epigenetics 2012, 7, 1258–1267. [Google Scholar] [CrossRef] [Green Version]
- Haber, D.A.; Velculescu, V.E. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Wei, M.; Grushko, T.A.; Dignam, J.; Hagos, F.; Nanda, R.; Sveen, L.; Xu, J.; Fackenthal, J.; Tretiakova, M.; Das, S.; et al. BRCA1 promoter methylation in sporadic breast cancer is associated with reduced BRCA1 copy number and chromosome 17 aneusomy. Cancer Res. 2005, 65, 10692–10699. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, A.M.; Spearman, A.D.; Tseng, S.Y.; Sweet, K.; Ewart Toland, A. Methylation not a frequent ‘‘second hit’’ in tumors with germline BRCA mutations. Fam. Cancer 2009, 8, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Morak, M.; Ibisler, A.; Keller, G.; Jessen, E.; Laner, A.; Gonzales-Fassrainer, D.; Locher, M.; Massdorf, T.; Nissen, A.M.; Benet-Pagès, A.; et al. Comprehensive analysis of the MLH1 promoter region in 480 patients with colorectal cancer and 1150 controls reveals new variants including one with a heritable constitutional MLH1 epimutation. J. Med. Genet. 2018, 55, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.R.; van Veen, E.M.; Byers, H.J.; Wallace, A.J.; Ellingford, J.M.; Beaman, G.; Santoyo-Lopez, J.; Aitman, T.J.; Eccles, D.M.; Lalloo, F.I.; et al. A Dominantly Inherited 5′ UTR Variant Causing Methylation-Associated Silencing of BRCA1 as a Cause of Breast and Ovarian Cancer. Am. J. Hum Genet. 2018, 103, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Böck, J.; Appenzeller, S.; Haertle, L.; Schneider, T.; Gehrig, A.; Schröder, J.; Rost, S.; Wolf, B.; Bartram, C.R.; Sutter, C.; et al. Single CpG hypermethylation, allele methylation errors, and decreased expression of multiple tumor suppressor genes in normal body cells of mutation-negative early-onset and high-risk breast cancer patients. Int. J. Cancer 2018, 143, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Wan, E.S.; Qiu, W.; Baccarelli, A.; Carey, V.J.; Bacherman, H.; Rennard, S.I.; Agusti, A.; Anderson, W.; Lomas, D.A.; Demeo, D.L. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum. Mol. Genet. 2012, 21, 3073–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela-Rey, M.; Woodhoo, A.; Martinez-Chantar, M.L.; Mato, J.M.; Lu, S.C. Alcohol; DNA methylation; and cancer. Alcohol Res. 2013, 35, 25–35. [Google Scholar]
- Ruiz-Hernandez, A.; Kuo, C.C.; Rentero-Garrido, P.; Tang, W.Y.; Redon, J.; Ordovas, J.M.; Navas-Acien, A.; Tellez-Plaza, M. Environmental chemicals and DNA methylation in adults: A systematic review of the epidemiologic evidence. Clin. Epigenet. 2015, 7, 55. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Wilson, A.; Koo, C.; Masrour, N.; Gallon, J.; Loomis, E.; Flower, K.; Wilhelm-Benartzi, C.; Hergovich, A.; Cunnea, P.; et al. Platinum-Based Chemotherapy Induces Methylation Changes in Blood DNA Associated with Overall Survival in Patients with Ovarian Cancer. Clin. Cancer Res. 2017, 23, 2213–2222. [Google Scholar] [CrossRef]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef] [Green Version]
- Barekati, Z.; Radpour, R.; Lu, Q.; Bitzer, J.; Zheng, H.; Toniolo, P.; Lenner, P.; Zhong, X.Y. Methylation signature of lymph node metastases in breast cancer patients. BMC Cancer 2012, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- Radpour, R.; Kohler, C.; Haghighi, M.M.; Fan, A.X.; Holzgreve, W.; Zhong, X.Y. Methylation profiles of 22 candidate genes in breast cancer using high-throughput MALDI-TOF mass array. Oncogene 2009, 28, 2969–2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, P.L.; Miron, A.; Andersen, L.M.; Iglehart, J.D.; Marks, J.R. Isolation and initial characterization of the BRCA2 promoter. Oncogene 1999, 18, 6000–6012. [Google Scholar] [CrossRef] [PubMed]
- Barekati, Z.; Radpour, R.; Kohler, C.; Zhang, B.; Toniolo, P.; Lenner, P.; Lv, Q.; Zheng, H.; Zhong, X.Y. Methylation profile of TP53 regulatory pathway and mtDNA alterations in breast cancer patients lacking TP53 mutations. Hum. Mol. Genet. 2010, 19, 2936–2946. [Google Scholar] [CrossRef]
- Caldeira, J.R.; Prando, E.C.; Quevedo, F.C.; Neto, F.A.; Rainho, C.A.; Rogatto, S.R. CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer 2006, 6, 48. [Google Scholar] [CrossRef]
- Esteller, M.; Avizienyte, E.; Corn, P.G.; Lothe, R.A.; Baylin, S.B.; Aaltonen, L.A.; Herman, J.G. Epigenetic inactivation of LKB1 in primary tumors associated with the Peutz-Jeghers syndrome. Oncogene 2000, 19, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Vo, Q.N.; Kim, W.J.; Cvitanovic, L.; Boudreau, D.A.; Ginzinger, D.G.; Brown, K.D. The ATM gene is a target for epigenetic silencing in locally advanced breast cancer. Oncogene 2004, 23, 9432–9437, Erratum in Oncogene 2005, 24, 1964. [Google Scholar] [CrossRef] [PubMed]
- Delmonico, L.; Moreira Ados, S.; Franco, M.F.; Esteves, E.B.; Scherrer, L.; Gallo, C.V.; do Nascimento, C.M.; Ornellas, M.H.; de Azevedo, C.M.; Alves, G. CDKN2A (p14(ARF)/p16(INK4a)) and ATM promoter methylation in patients with impalpable breast lesions. Hum. Pathol. 2015, 46, 1540–1547. [Google Scholar] [CrossRef]
- Watanabe, Y.; Maeda, I.; Oikawa, R.; Wu, W.; Tsuchiya, K.; Miyoshi, Y.; Itoh, F.; Tsugawa, K.; Ohta, T. Aberrant DNA methylation status of DNA repair genes in breast cancer treated with neoadjuvant chemotherapy. Genes Cells. 2013, 18, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Lao, V.V.; Welcsh, P.; Luo, Y.; Carter, K.T.; Dzieciatkowski, S.; Dintzis, S.; Meza, J.; Sarvetnick, N.E.; Monnat, R.J., Jr.; Loeb, L.A.; et al. Altered RECQ Helicase Expression in Sporadic Primary Colorectal Cancers. Transl. Oncol. 2013, 6, 458–469. [Google Scholar] [CrossRef]
- Meier, D.; Schindler, D. Fanconi anemia core complex gene promoters harbor conserved transcription regulatory elements. PLoS ONE 2011, 6, e22911. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Bedeschi, M.F.; Maitz, S.; Cereda, A.; Faré, C.; Motta, S.; Seresini, A.; D’Ursi, P.; Orro, A.; Pecile, V.; et al. Characterization of multi-locus imprinting disturbances and underlying genetic defects in patients with chromosome 11p15.5 related imprinting disorders. Epigenetics 2018, 13, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Pizzamiglio, S.; Verderio, P.; Marubini, E. Bootstrap confidence intervals for nucleic acid concentration by absolute real-time PCR. BioMed. Stat. Clin. Epidemiol. 2008, 2, 109–115. [Google Scholar]
- Hosmer, D.W.; Lemeshow, S. Applied Logistic Regression, 3rd ed.; John Wiley & Sons: New York, NY, USA, 1989; pp. 47–77. [Google Scholar]
- Artusi, R.; Verderio, P.; Marubini, E. Bravais-Pearson and Spearman correlation coefficients: Meaning, test of hypothesis and confidence interval. Int. J. Biol. Mark. 2002, 17, 148–151. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breast Cancer Histotype | Ductal | Lobular | Mixed | Other | n.a. | Total | |
|---|---|---|---|---|---|---|---|
| Number of cases | 129 | 2 | 4 | 8 | 11 | 154 | |
| Median Age at diagnosis (range) | 32 (19–44) | 30; 35 | 30 (24–35) | 34.5 (26–38) | 34 (31–40) | 33 (19–44) | |
| Grade | I | 10 (7.7%) | - | - | - | - | 10 (6.5%) |
| II | 38 (29.5%) | 2 (100%) | 3 (75%) | 2 (25%) | 2 (18.2%) | 47 (30.5%) | |
| III | 68 (52.7%) | - | 1 (25%) | 5 (62.5%) | 6 (54.5%) | 80 (51.9%) | |
| n.a. | 13 (10.1%) | - | - | 1 (12.5%) | 3 (27.3%) | 17 (11.0%) | |
| Tumor size (pT) | Is | 4 (3.1%) | - | - | - | - | 4 (2.6%) |
| 1 | 67 (51.9%) | - | - | 3 (37.5%) | - | 70 (45.5%) | |
| 2 | 30 (23.3%) | 1 (50%) | 3 (75%) | 4 (50%) | 3 (27.3%) | 41 (26.6%) | |
| 3 | 3 (2.3%) | - | - | - | - | 3 (1.9%) | |
| 4 | 2 (1.6%) | 1 (50%) | - | - | - | 3 (1.9%) | |
| n.a. | 23 (17.8%) | - | 1 (25%) | 1 (12.5%) | 8 (72.7%) | 33 (21.4%) | |
| Lymph Node Metastasis (pN) | + | 57 (44.2%) | - | 3 (75%) | 3 (37.5%) | 6 (54.5%) | 69 (44.8%) |
| − | 63 (48.8) | 2 (100%) | 1 (25%) | 5 (62.5%) | 2 (18.2%) | 73 (47.4%) | |
| n.a. | 9 (7%) | - | - | - | 3 (27.3%) | 12 (7.8%) | |
| Distant Metastasis (M) | + | 8 (6.2%) | - | 1 (25%) | 2 (25%) | 1 (9.1) | 12 (7.8%) |
| − | 121 (93.8%) | 2 (100%) | 3 (75%) | 6 (75%) | 10 (90.9%) | 142 (92.2%) | |
| Estrogen Receptor (ER staining) | + | 80 (62%) | 1 (50%) | 4 (100%) | 5 (62.5%) | 5 (45.4%) | 95 (61.7%) |
| − | 45 (34.9%) | 1(50%) | - | 3 (37.5%) | 4 (36.4%) | 53 (34.4%) | |
| n.a. | 4 (3.1%) | - | - | - | 2 (18.2%) | 6 (3.9%) | |
| Progesterone Receptor (PgR staining) | + | 57 (44.2%) | 2 (100%) | 4 (100%) | 4 (50%) | 5 (45.4%) | 72 (46.8%) |
| − | 65 (50.4%) | - | - | 4 (50%) | 4 (36.4%) | 73 (47.4%) | |
| n.a. | 7 (5.4%) | - | - | - | 2 (18.2%) | 9 (5.8%) | |
| HER2 staining | + | 45 (34.9%) | 1 (50%) | - | - | 1 (9.1%) | 47 (30.5%) |
| − | 56 (43.4%) | - | 1 (25%) | 6 (75%) | 7 (63.6%) | 70 (45.5%) | |
| n.a. | 28 (21.7%) | 1 (50%) | 3 (75%) | 2 (25%) | 3 (27.3%) | 37 (24.0%) | |
| Triple Negative Phenotype | 25 (19.4%) | - | - | 3 (37.5%) | 3 (27.3%) | 31 (20.1%) | |
| 2nd Breast Cancer | 17 (13.2%) | 1 (50%) | 2 (50%) | - | 1 (9.1%) | 21 (13.6%) | |
| 2nd Breast Cancer Median age (range) | 42 (29–68) | 38 | 50;33 | - | 38 | 42 (29–68) | |
| Blood withdrawal (before/after chemotherap) | before CT | 37 (28.7%) | - | - | 1 (12.5%) | - | 38 (24.7%) |
| after CT | 92 (71.3%) | 2 (100%) | 4 (100%) | 7 (87.5%) | 11 (100%) | 116 (75.3%) | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzollini, J.; Pesenti, C.; Pizzamiglio, S.; Fontana, L.; Guarino, C.; Peissel, B.; Plebani, M.; Tabano, S.; Sirchia, S.M.; Colapietro, P.; et al. Constitutive BRCA1 Promoter Hypermethylation Can Be a Predisposing Event in Isolated Early-Onset Breast Cancer. Cancers 2019, 11, 58. https://doi.org/10.3390/cancers11010058
Azzollini J, Pesenti C, Pizzamiglio S, Fontana L, Guarino C, Peissel B, Plebani M, Tabano S, Sirchia SM, Colapietro P, et al. Constitutive BRCA1 Promoter Hypermethylation Can Be a Predisposing Event in Isolated Early-Onset Breast Cancer. Cancers. 2019; 11(1):58. https://doi.org/10.3390/cancers11010058
Chicago/Turabian StyleAzzollini, Jacopo, Chiara Pesenti, Sara Pizzamiglio, Laura Fontana, Carmela Guarino, Bernard Peissel, Maddalena Plebani, Silvia Tabano, Silvia Maria Sirchia, Patrizia Colapietro, and et al. 2019. "Constitutive BRCA1 Promoter Hypermethylation Can Be a Predisposing Event in Isolated Early-Onset Breast Cancer" Cancers 11, no. 1: 58. https://doi.org/10.3390/cancers11010058
APA StyleAzzollini, J., Pesenti, C., Pizzamiglio, S., Fontana, L., Guarino, C., Peissel, B., Plebani, M., Tabano, S., Sirchia, S. M., Colapietro, P., Villa, R., Paolini, B., Verderio, P., Miozzo, M., & Manoukian, S. (2019). Constitutive BRCA1 Promoter Hypermethylation Can Be a Predisposing Event in Isolated Early-Onset Breast Cancer. Cancers, 11(1), 58. https://doi.org/10.3390/cancers11010058