Bortezomib Augments Natural Killer Cell Targeting of Stem-Like Tumor Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
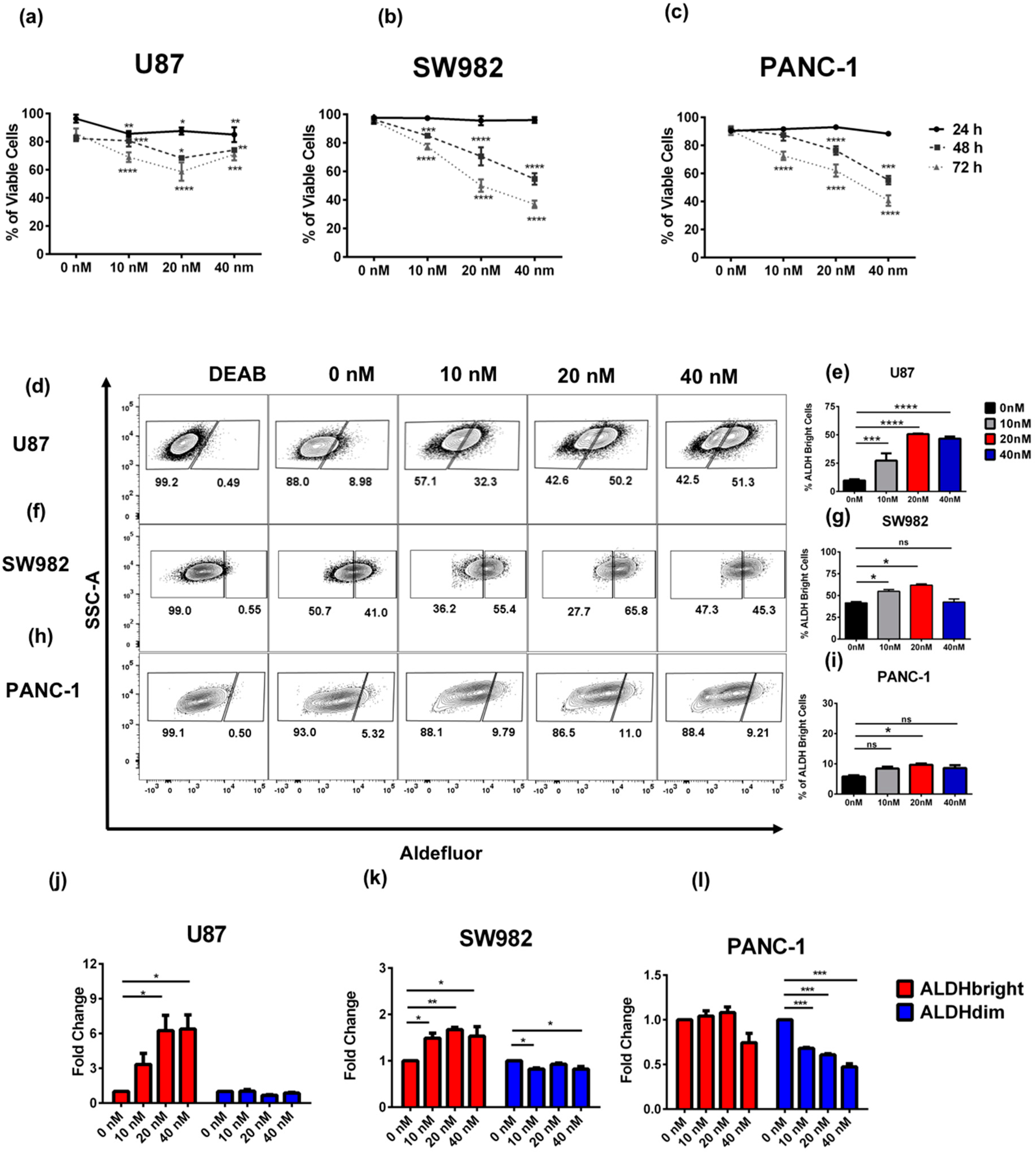
2.1. Bortezomib Treatment Enriches for ALDHbright Tumor Cells from Several Solid Tumor Types
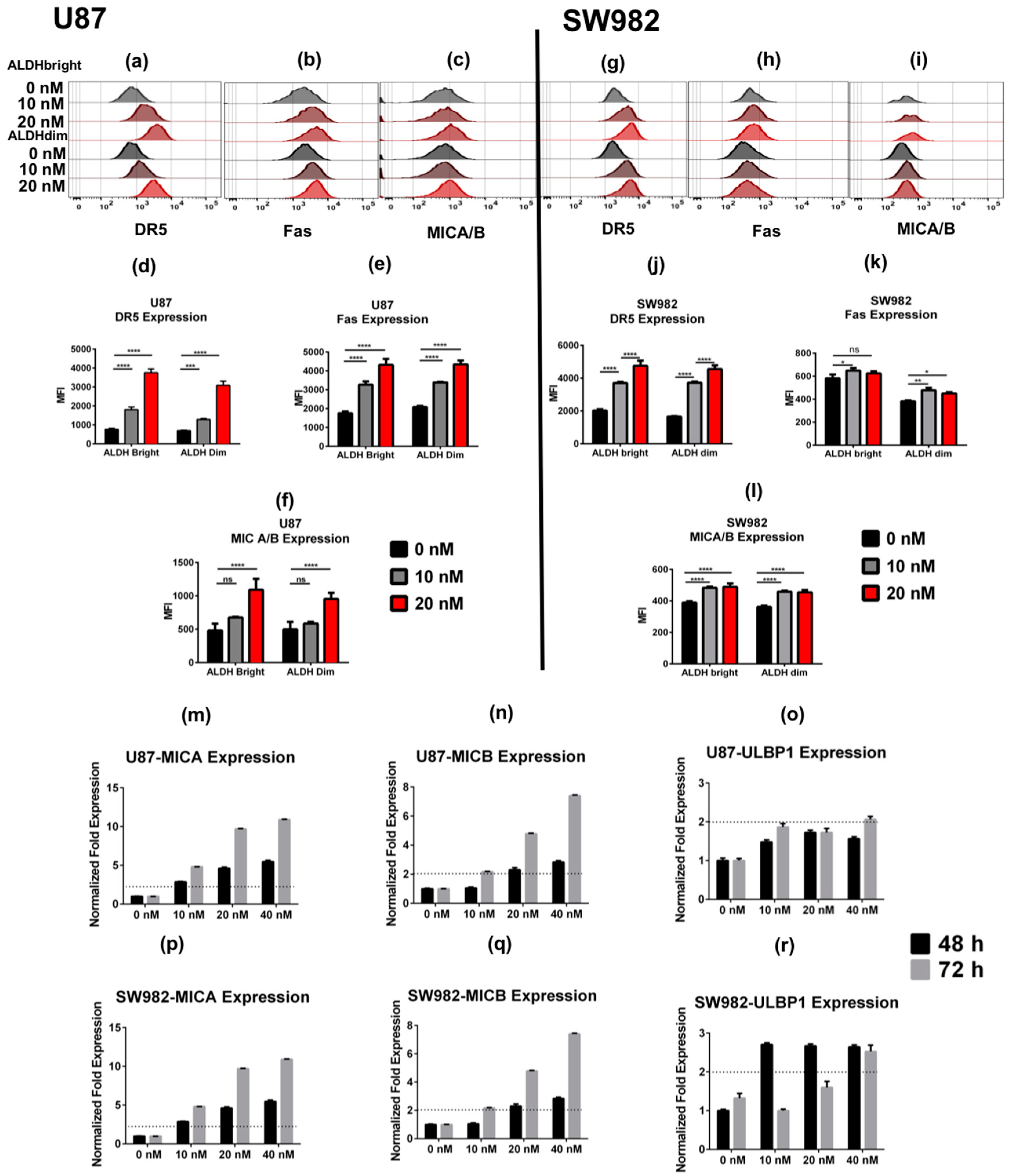
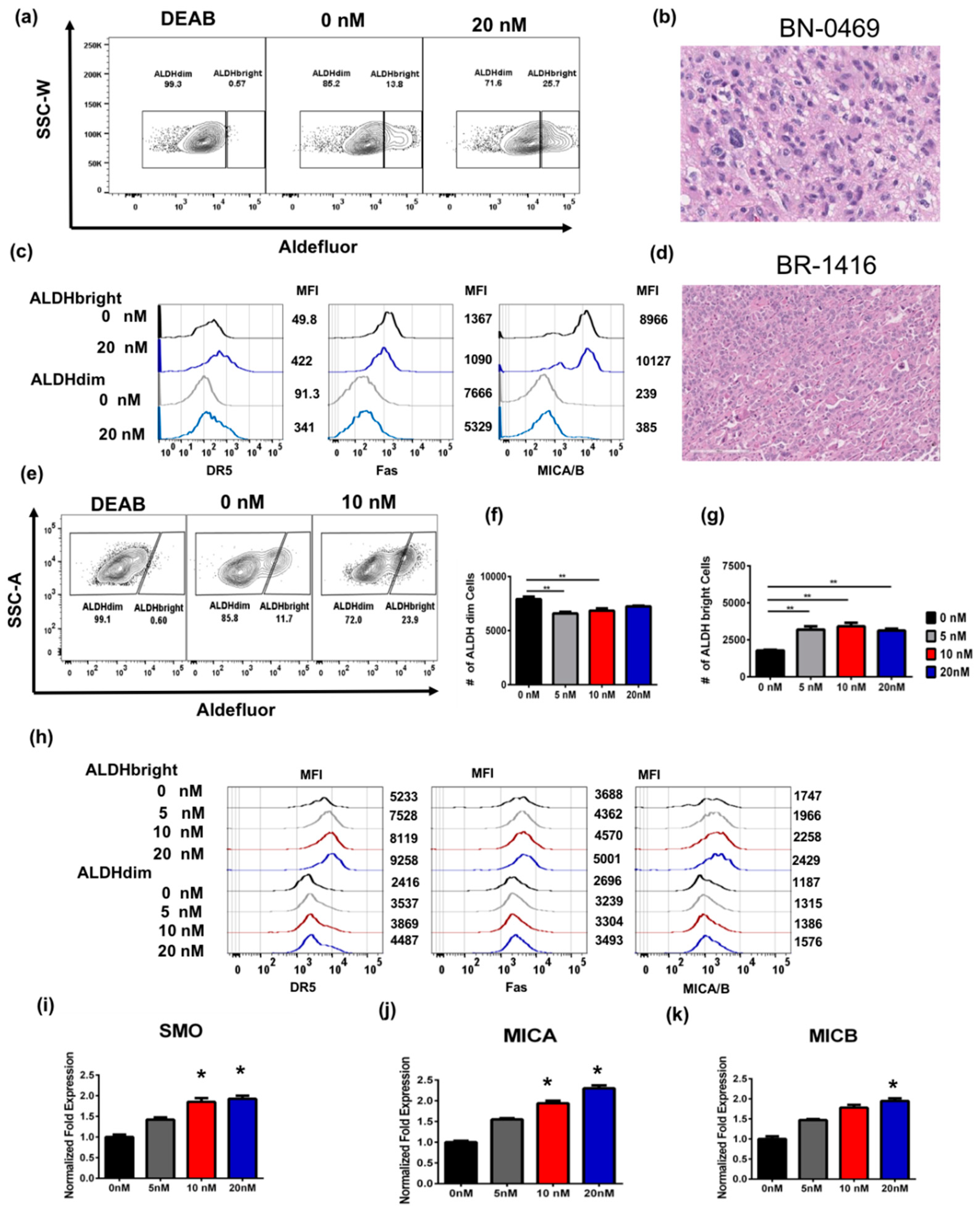
2.2. Bortezomib Increases the Expression of Stress Ligands and Death Receptors on both ALDHbright and ALDHdim Cells
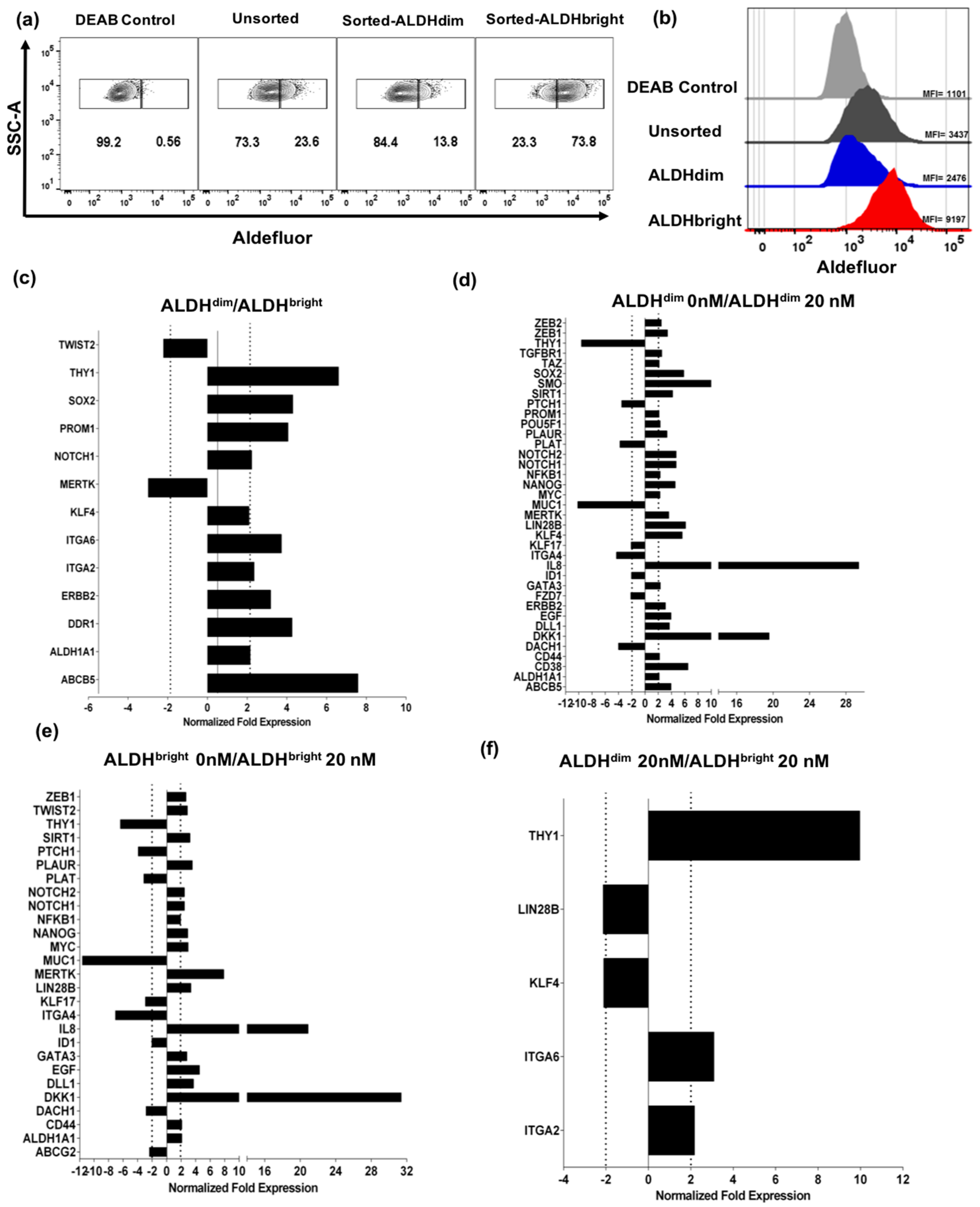
2.3. U87 ALDHbright Subpopulations Exhibit Increased Expression of Cancer Stem Cell-Related Markers Compared to ALDHdim Subpopulations
Bortezomib Affects Expression of Cancer Stem Cell-Related Genes and MICA/B in both ALDHbright and ALDHdim Subpopulations
2.4. Bortezomib Enriches for ALDHbright Cells from Human Primary Glioblastoma and Breast Tumor
2.5. ALDHbright Cells Are Sensitized to NK Cell Killing following Bortezomib Pre-Treatment In Vitro
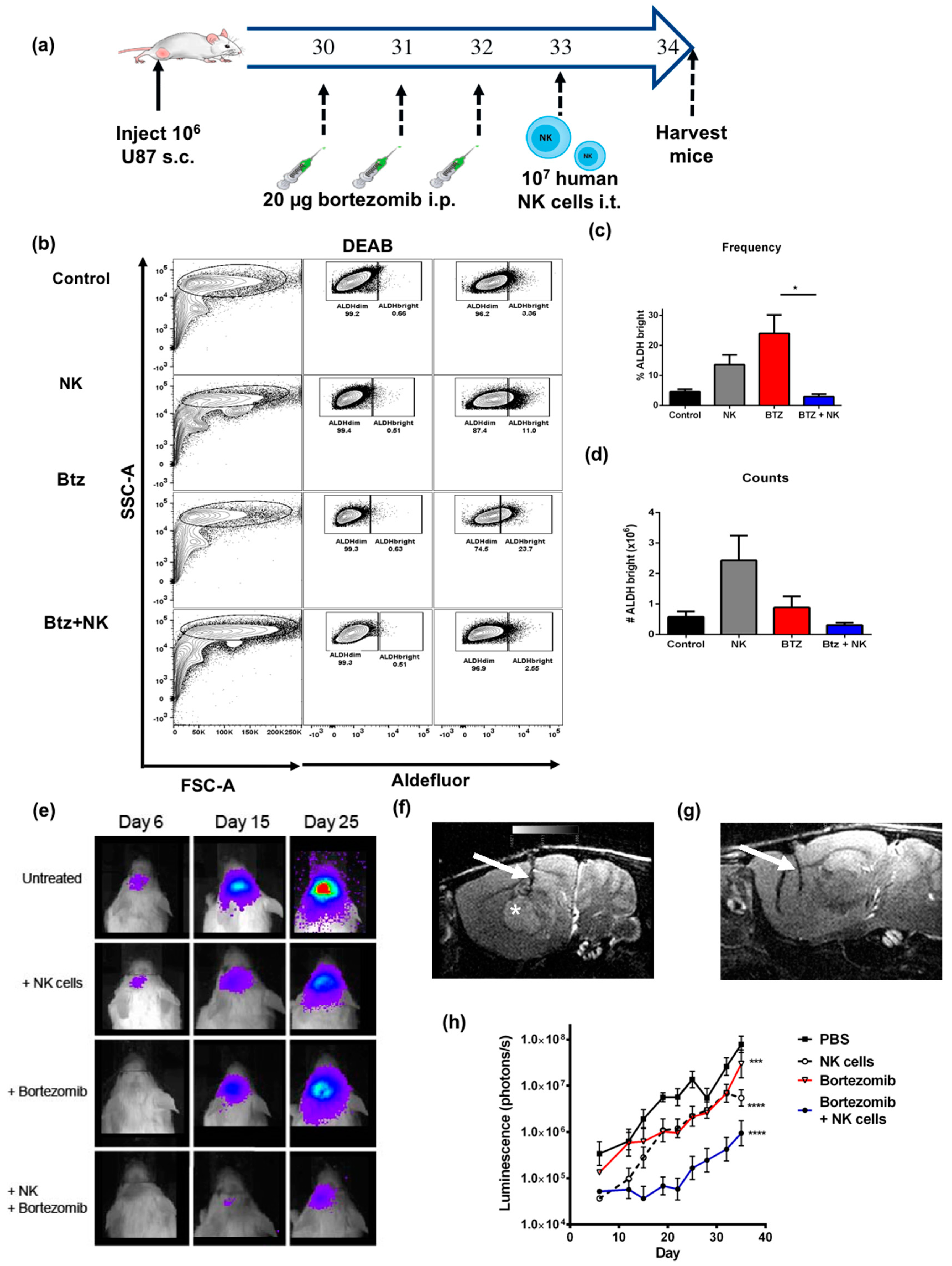
2.6. The Combination of NK Cells and Bortezomib Leads to Reduced Stem-Like Tumor Cells In Vivo and Promotes Tumor Regression
3. Discussion
4. Materials and Methods
4.1. Tumor Cell Lines
4.2. NK Cell Isolation
4.3. NK Cell Expansion and Activation
4.4. Bortezomib Treatment Assays
4.5. Flow Cytometric Cytotoxicity Assay
4.6. Flow Cytometry and Cell Sorting
4.7. Tumor Models
4.8. Hydrodynamic IL-15 Plasmid Delivery
4.9. In Vivo Imaging
4.10. RNA Extraction and qPCR
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALDH | Aldehyde dehydrogenase |
| CSC | Cancer stem cell |
| NK | Natural killer |
| DR5 | Death receptor 5 |
| MIC A/B | MHC class I polypeptide-related sequence A/B |
References
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.I.; Grossenbacher, S.K.; Murphy, W.J.; Canter, R.J. Targeting Cancer Stem Cells with Natural Killer Cell Immunotherapy. Expert Opin. Biol. Ther. 2017, 17, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chai, S.; Wang, P.; Zhang, C.; Yang, Y.; Yang, Y.; Wang, K. Aldehyde dehydrogenases and cancer stem cells. Cancer Lett. 2015, 369, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Ma, I.; Allan, A.L. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. 2011, 7, 292–306. [Google Scholar] [CrossRef]
- Deng, S.; Yang, X.; Lassus, H.; Liang, S.; Kaur, S.; Ye, Q.; Li, C.; Wang, L.P.; Roby, K.F.; Orsulic, S.; et al. Distinct expression levels and patterns of stem cell marker, aldehyde dehydrogenase isoform 1 (ALDH1), in human epithelial cancers. PLoS ONE 2010, 5, e10277. [Google Scholar] [CrossRef]
- Li, T.; Su, Y.; Mei, Y.; Leng, Q.; Leng, B.; Liu, Z.; Stass, S.A.; Jiang, F. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Investig. 2010, 90, 234–244. [Google Scholar] [CrossRef]
- Lingala, S.; Cui, Y.Y.; Chen, X.; Ruebner, B.H.; Qian, X.F.; Zern, M.A.; Wu, J. Immunohistochemical staining of cancer stem cell markers in hepatocellular carcinoma. Exp. Mol. Pathol. 2010, 89, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, Z.A.; Yang, J.; Wang, Q.; Kowalski, J.; Freed, I.; Murter, C.; Hong, S.M.; Koorstra, J.B.; Rajeshkumar, N.V.; He, X.; et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J. Natl. Cancer Inst. 2010, 102, 340–351. [Google Scholar] [CrossRef]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Ucar, D.; Cogle, C.R.; Zucali, J.R.; Ostmark, B.; Scott, E.W.; Zori, R.; Gray, B.A.; Moreb, J.S. Aldehyde dehydrogenase activity as a functional marker for lung cancer. Chem. Biol. Interact. 2009, 178, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wan, L.; Geng, J.; Wu, C.L.; Bai, X. Aldehyde dehydrogenase 1A1 possesses stem-like properties and predicts lung cancer patient outcome. J. Thorac. Oncol. 2012, 7, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Sophos, N.A.; Pappa, A.; Ziegler, T.L.; Vasiliou, V. Aldehyde dehydrogenase gene superfamily: The 2000 update. Chem. Biol. Interact. 2001, 130–132, 323–337. [Google Scholar] [CrossRef]
- Jackson, B.; Brocker, C.; Thompson, D.C.; Black, W.; Vasiliou, K.; Nebert, D.W.; Vasiliou, V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genom. 2011, 5, 283–303. [Google Scholar] [CrossRef]
- Ames, E.; Canter, R.J.; Grossenbacher, S.K.; Mac, S.; Chen, M.; Smith, R.C.; Hagino, T.; Perez-Cunningham, J.; Sckisel, G.D.; Urayama, S.; et al. NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. J. Immunol. 2015, 195, 4010–4019. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Vivier, E.; Nunes, J.A.; Vely, F. Natural killer cell signaling pathways. Science 2004, 306, 1517–1519. [Google Scholar] [CrossRef]
- Tallerico, R.; Garofalo, C.; Carbone, E. A New Biological Feature of Natural Killer Cells: The Recognition of Solid Tumor-Derived Cancer Stem Cells. Front. Immunol. 2016, 7, 179. [Google Scholar] [CrossRef]
- Ames, E.; Hallett, W.H.; Murphy, W.J. Sensitization of human breast cancer cells to natural killer cell-mediated cytotoxicity by proteasome inhibition. Clin. Exp. Immunol. 2009, 155, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Hallett, W.H.; Ames, E.; Motarjemi, M.; Barao, I.; Shanker, A.; Tamang, D.L.; Sayers, T.J.; Hudig, D.; Murphy, W.J. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J. Immunol. 2008, 180, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yue, P.; Chen, S.; Hu, L.; Lonial, S.; Khuri, F.R.; Sun, S.Y. The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 2007, 67, 4981–4988. [Google Scholar] [CrossRef] [PubMed]
- Armeanu, S.; Krusch, M.; Baltz, K.M.; Weiss, T.S.; Smirnow, I.; Steinle, A.; Lauer, U.M.; Bitzer, M.; Salih, H.R. Direct and natural killer cell-mediated antitumor effects of low-dose bortezomib in hepatocellular carcinoma. Clin. Cancer Res. 2008, 14, 3520–3528. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, A.; Berg, M.; Smith, A.; Childs, R.W. Bortezomib Treatment to Potentiate the Anti-tumor Immunity of Ex-vivo Expanded Adoptively Infused Autologous Natural Killer Cells. J. Cancer 2011, 2, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, A.; Abrams, S.I.; Schrump, D.S.; Alvarez, G.; Suffredini, D.; Berg, M.; Childs, R. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: A novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006, 66, 7317–7325. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Tricot, G.J.; Garg, T.K.; Malaviarachchi, P.A.; Szmania, S.M.; Kellum, R.E.; Storrie, B.; Mulder, A.; Shaughnessy, J.D., Jr.; Barlogie, B.; et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood 2008, 111, 1309–1317. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Abou-Antoun, T.J.; Hale, J.S.; Lathia, J.D.; Dombrowski, S.M. Brain Cancer Stem Cells in Adults and Children: Cell Biology and Therapeutic Implications. Neurother. J. Am. Soc. Expe. Neurother. 2017, 14, 372–384. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Liu, Y.; Zhu, T.; Zhu, J.; Dimeco, F.; Vescovi, A.L.; Heth, J.A.; Muraszko, K.M.; Fan, X.; Lubman, D.M. CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Mol. Cell. Proteom. MCP 2012, 11, 010744. [Google Scholar] [CrossRef]
- Krebsbach, P.H.; Villa-Diaz, L.G. The Role of Integrin alpha6 (CD49f) in Stem Cells: More than a Conserved Biomarker. Stem Cells Dev. 2017, 26, 1090–1099. [Google Scholar] [CrossRef]
- Infante, P.; Alfonsi, R.; Ingallina, C.; Quaglio, D.; Ghirga, F.; D’Acquarica, I.; Bernardi, F.; Di Magno, L.; Canettieri, G.; Screpanti, I.; et al. Inhibition of Hedgehog-dependent tumors and cancer stem cells by a newly identified naturally occurring chemotype. Cell Death Dis. 2016, 7, e2376. [Google Scholar] [CrossRef] [PubMed]
- Canter, R.J.; Grossenbacher, S.K.; Foltz, J.A.; Sturgill, I.R.; Park, J.S.; Luna, J.I.; Kent, M.S.; Culp, W.T.N.; Chen, M.; Modiano, J.F.; et al. Radiotherapy enhances natural killer cell cytotoxicity and localization in pre-clinical canine sarcomas and first-in-dog clinical trial. J. Immunother. Cancer 2017, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Withers, S.S.; Modiano, J.F.; Kent, M.S.; Chen, M.; Luna, J.I.; Culp, W.T.N.; Sparger, E.E.; Rebhun, R.B.; Monjazeb, A.M.; et al. Canine cancer immunotherapy studies: Linking mouse and human. J. Immunother. Cancer 2016, 4, 97. [Google Scholar] [CrossRef] [PubMed]
- Ames, E.; Canter, R.J.; Grossenbacher, S.K.; Mac, S.; Smith, R.C.; Monjazeb, A.M.; Chen, M.; Murphy, W.J. Enhanced targeting of stem-like solid tumor cells with radiation and natural killer cells. Oncoimmunology 2015, 4, e1036212. [Google Scholar] [CrossRef] [PubMed]
- Esquivel-Velazquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lauzon, W.; Lemaire, I. Alveolar macrophage inhibition of lung-associated NK activity: Involvement of prostaglandins and transforming growth factor-beta 1. Exp. Lung Res. 1994, 20, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Hentges, F.; Zimmer, J. Consequences of the crosstalk between monocytes/macrophages and natural killer cells. Front. Immunol. 2012, 3, 403. [Google Scholar] [CrossRef] [PubMed]
- Nunez, S.Y.; Ziblat, A.; Secchiari, F.; Torres, N.I.; Sierra, J.M.; Raffo Iraolagoitia, X.L.; Araya, R.E.; Domaica, C.I.; Fuertes, M.B.; Zwirner, N.W. Human M2 Macrophages Limit NK Cell Effector Functions through Secretion of TGF-beta and Engagement of CD85j. J. Immunol. 2018, 200, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Szkaradkiewicz, A.; Karpinski, T.M.; Drews, M.; Borejsza-Wysocki, M.; Majewski, P.; Andrzejewska, E. Natural killer cell cytotoxicity and immunosuppressive cytokines (IL-10, TGF-beta1) in patients with gastric cancer. J. Biomed. Biotechnol. 2010, 2010, 901564. [Google Scholar] [CrossRef] [PubMed]
- Woodward, W.A.; Chen, M.S.; Behbod, F.; Alfaro, M.P.; Buchholz, T.A.; Rosen, J.M. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 618–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullah, L.N.; Chow, E.K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, H.C.; Risau, W.; Konerding, M.A. Inhibition of proteasome function induces programmed cell death in proliferating endothelial cells. FASEB J. 2000, 14, 65–77. [Google Scholar] [CrossRef]
- Drexler, H.C. Activation of the cell death program by inhibition of proteasome function. Proc. Natl. Acad. Sci. USA 1997, 94, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef] [Green Version]
- Kremer, V.; Ligtenberg, M.A.; Zendehdel, R.; Seitz, C.; Duivenvoorden, A.; Wennerberg, E.; Colon, E.; Scherman-Plogell, A.H.; Lundqvist, A. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J. Immunother. Cancer 2017, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, C.; Huergo-Zapico, L.; Parodi, M.; Pedrazzi, M.; Mingari, M.C.; Moretta, A.; Sparatore, B.; Gonzalez, S.; Olive, D.; Bottino, C.; et al. NK Cells, Tumor Cell Transition, and Tumor Progression in Solid Malignancies: New Hints for NK-Based Immunotherapy? J. Immunol. Res. 2016, 2016, 4684268. [Google Scholar] [CrossRef]
- Wennerberg, E.; Kremer, V.; Childs, R.; Lundqvist, A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol. Immunother. CII 2015, 64, 225–235. [Google Scholar] [CrossRef]
- Somanchi, S.S.; Senyukov, V.V.; Denman, C.J.; Lee, D.A. Expansion, purification, and functional assessment of human peripheral blood NK cells. J. Vis. Exp. 2011. [Google Scholar] [CrossRef]
- Barao, I.; Alvarez, M.; Redelman, D.; Weiss, J.M.; Ortaldo, J.R.; Wiltrout, R.H.; Murphy, W.J. Hydrodynamic delivery of human IL-15 cDNA increases murine natural killer cell recovery after syngeneic bone marrow transplantation. Biol. Blood Marrow Transplant. 2011, 17, 1754–1764. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luna, J.I.; Grossenbacher, S.K.; Sturgill, I.R.; Ames, E.; Judge, S.J.; Bouzid, L.A.; Darrow, M.A.; Murphy, W.J.; Canter, R.J. Bortezomib Augments Natural Killer Cell Targeting of Stem-Like Tumor Cells. Cancers 2019, 11, 85. https://doi.org/10.3390/cancers11010085
Luna JI, Grossenbacher SK, Sturgill IR, Ames E, Judge SJ, Bouzid LA, Darrow MA, Murphy WJ, Canter RJ. Bortezomib Augments Natural Killer Cell Targeting of Stem-Like Tumor Cells. Cancers. 2019; 11(1):85. https://doi.org/10.3390/cancers11010085
Chicago/Turabian StyleLuna, Jesus I., Steven K. Grossenbacher, Ian R. Sturgill, Erik Ames, Sean J. Judge, Lyes A. Bouzid, Morgan A. Darrow, William J. Murphy, and Robert J. Canter. 2019. "Bortezomib Augments Natural Killer Cell Targeting of Stem-Like Tumor Cells" Cancers 11, no. 1: 85. https://doi.org/10.3390/cancers11010085
APA StyleLuna, J. I., Grossenbacher, S. K., Sturgill, I. R., Ames, E., Judge, S. J., Bouzid, L. A., Darrow, M. A., Murphy, W. J., & Canter, R. J. (2019). Bortezomib Augments Natural Killer Cell Targeting of Stem-Like Tumor Cells. Cancers, 11(1), 85. https://doi.org/10.3390/cancers11010085