A Novel Multi-Target Small Molecule, LCC-09, Inhibits Stemness and Therapy-Resistant Phenotypes of Glioblastoma Cells by Increasing miR-34a and Deregulating the DRD4/Akt/mTOR Signaling Axis


 , , ,
, , , 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs and Chemicals
2.2. Cell Lines and Culture
3. Western Blot Analysis
3.1. miR-34a Transfection
3.2. RNA Extraction and RT-PCR
3.3. Sulforhodamine B (SRB) Cell Viability Assay
3.4. Combination Median Effect Analysis
3.5. Neurosphere Formation Assay
3.6. Colony Formation Assay
3.7. Scratch Wound-Healing Migration Assay
3.8. Fluorescence-Activated Cell Sorting (FACS)
3.9. ALDEFLUOR ALDH Activity Analysis
3.10. Animal Studies
3.11. Statistical Analysis
4. Results
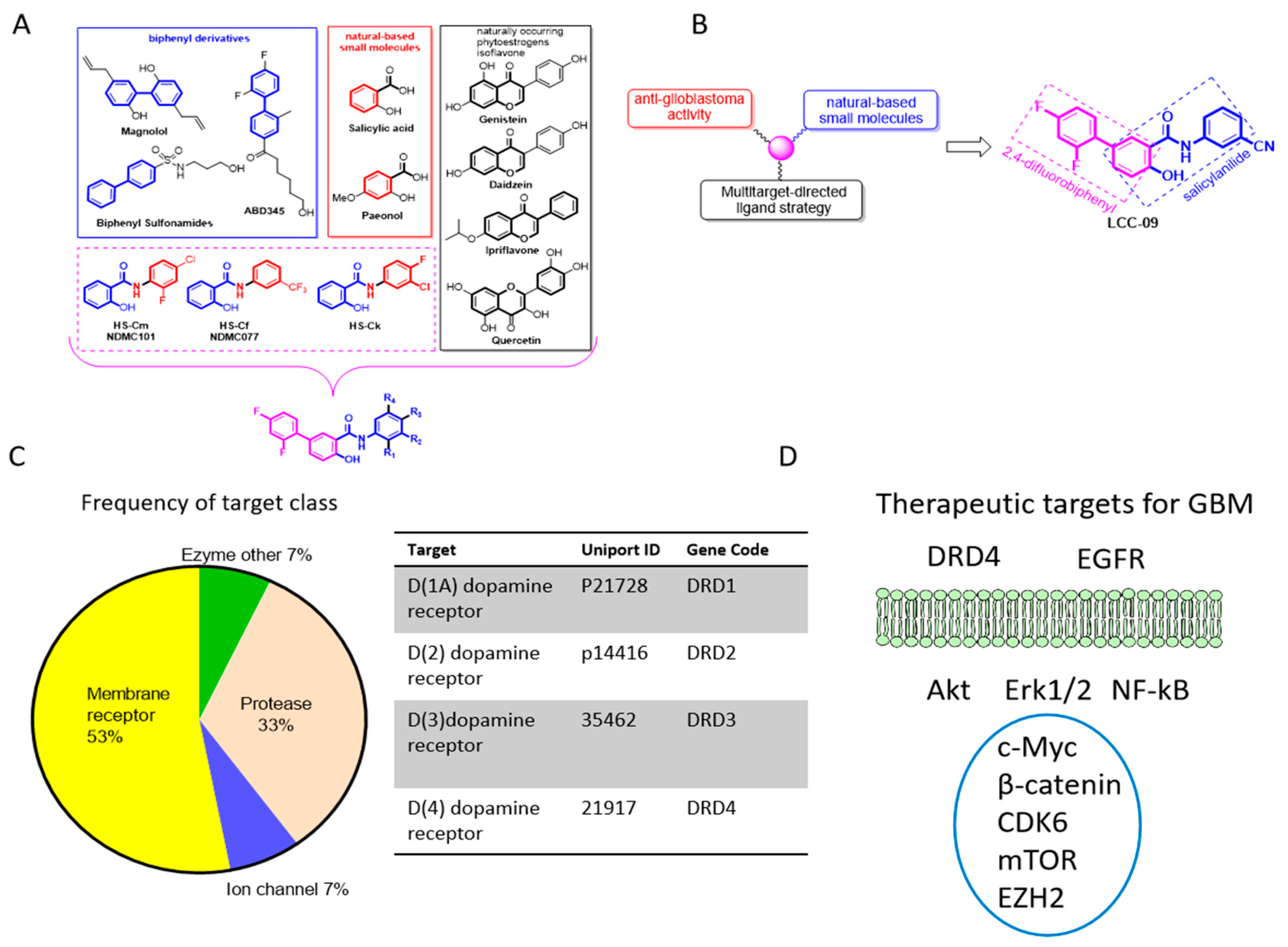
4.1. The Design, Synthesis and Functional Characterization of the Novel Multi-Target Small Molecule Dopamine Antagonist, Lcc-09
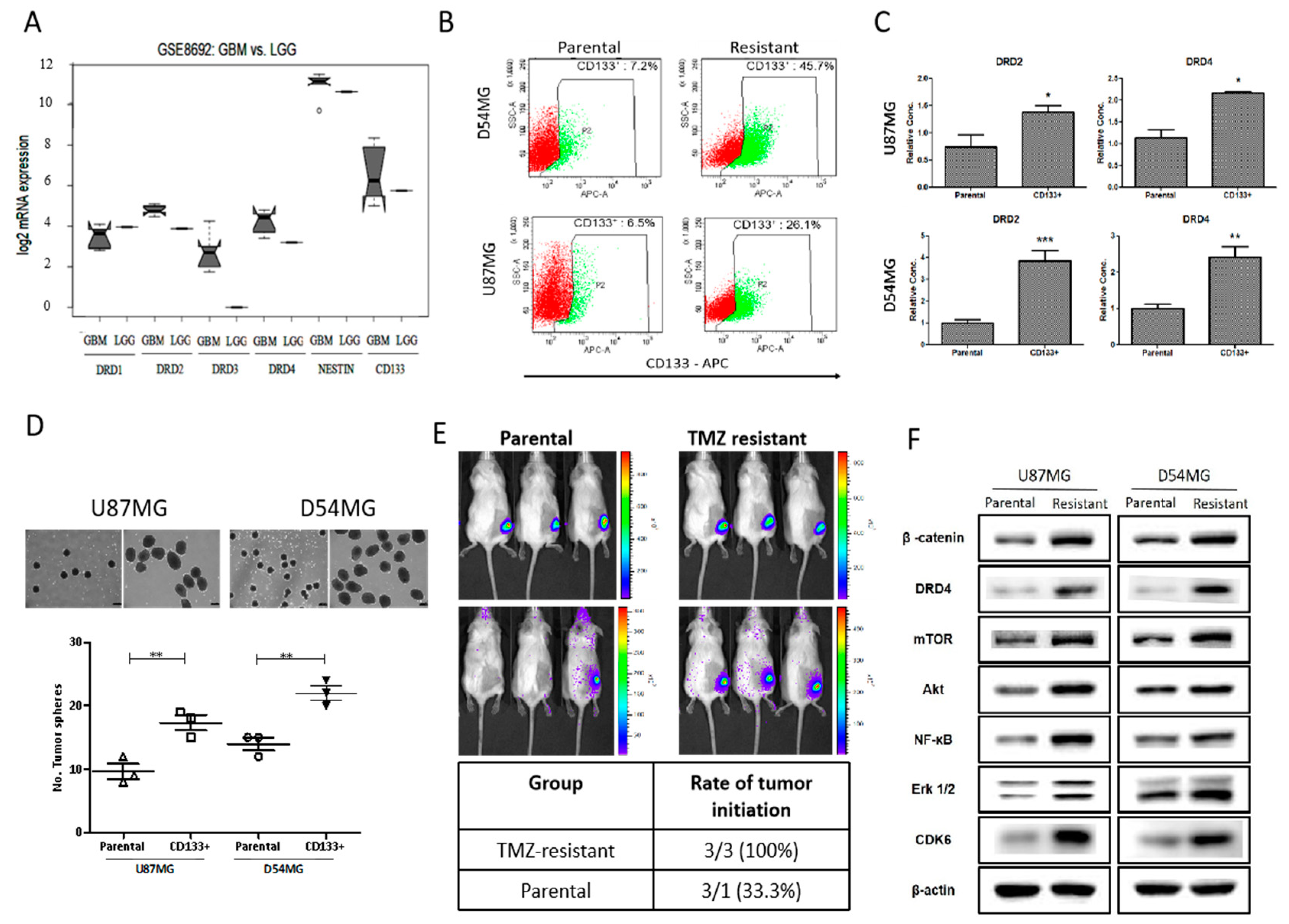
4.2. Enhanced cd133 Positivity and Constitutive drd4 Dopaminergic Signaling Characterize TMZ Resistance in gbm Cells
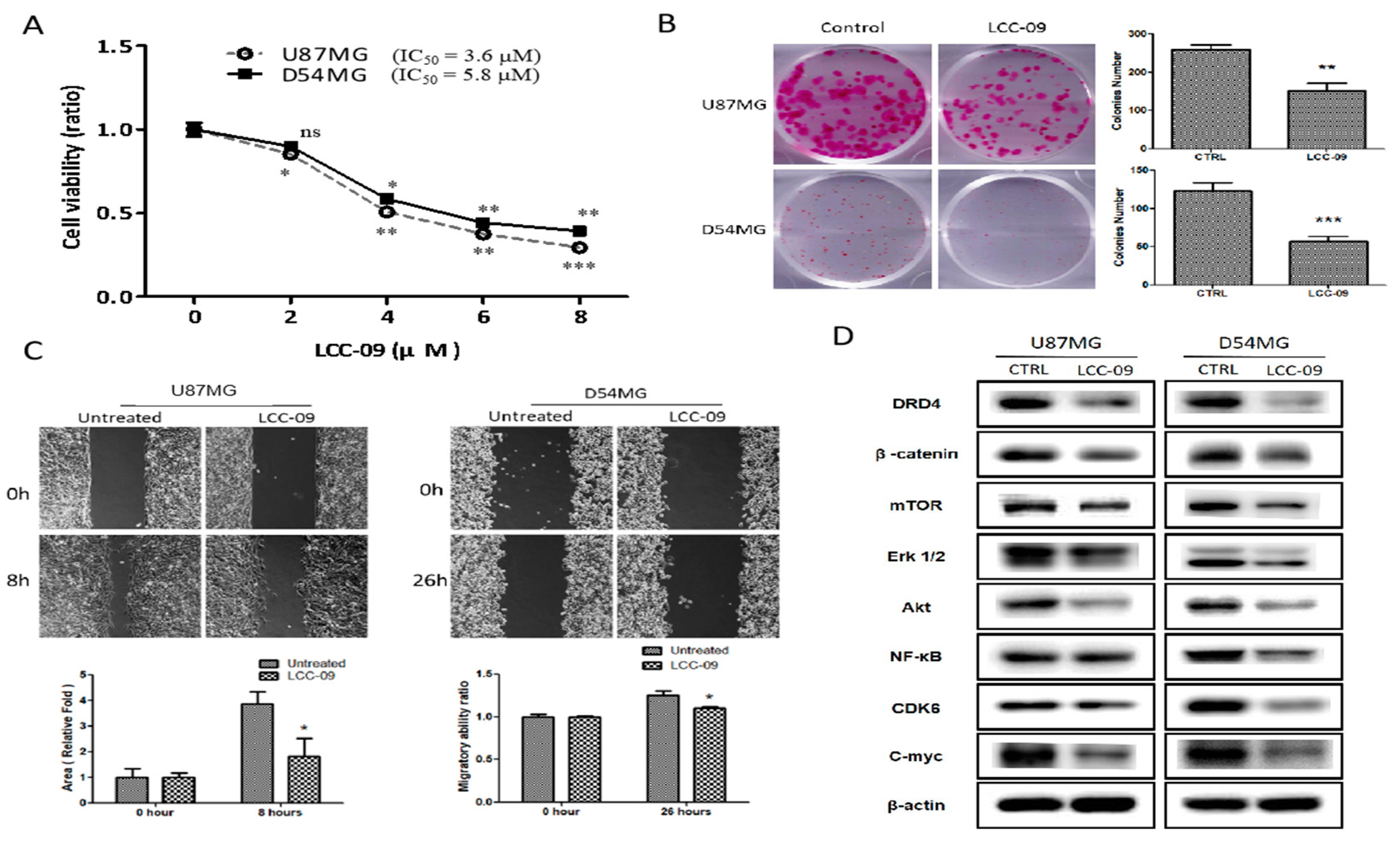
4.3. LCC-09 Suppressed the Viability and Metastatic Phenotype of GBM Cells through the Dysregulation of DRD4-Mediated AKT/mTOR and NF-κB Signaling Axes
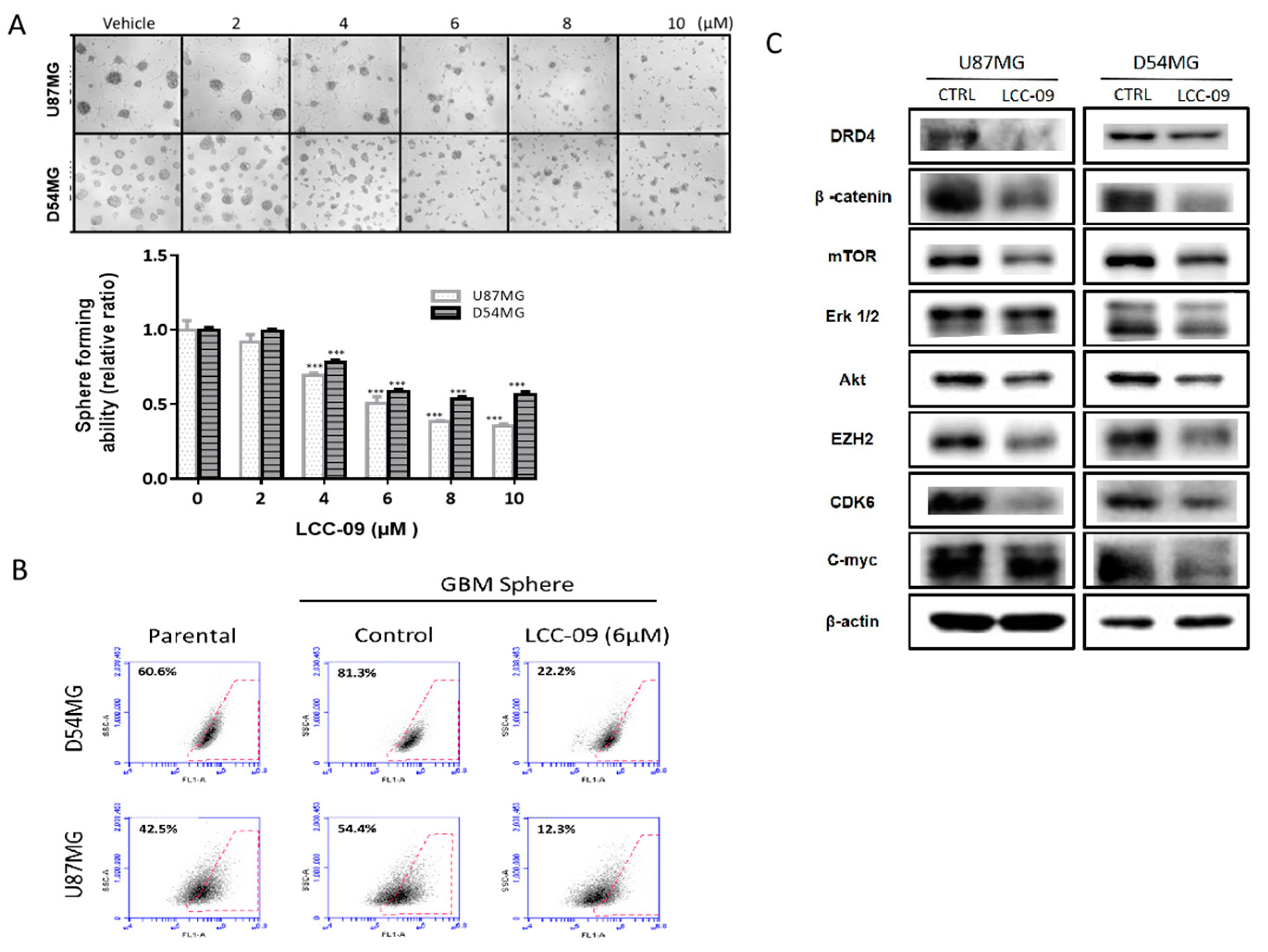
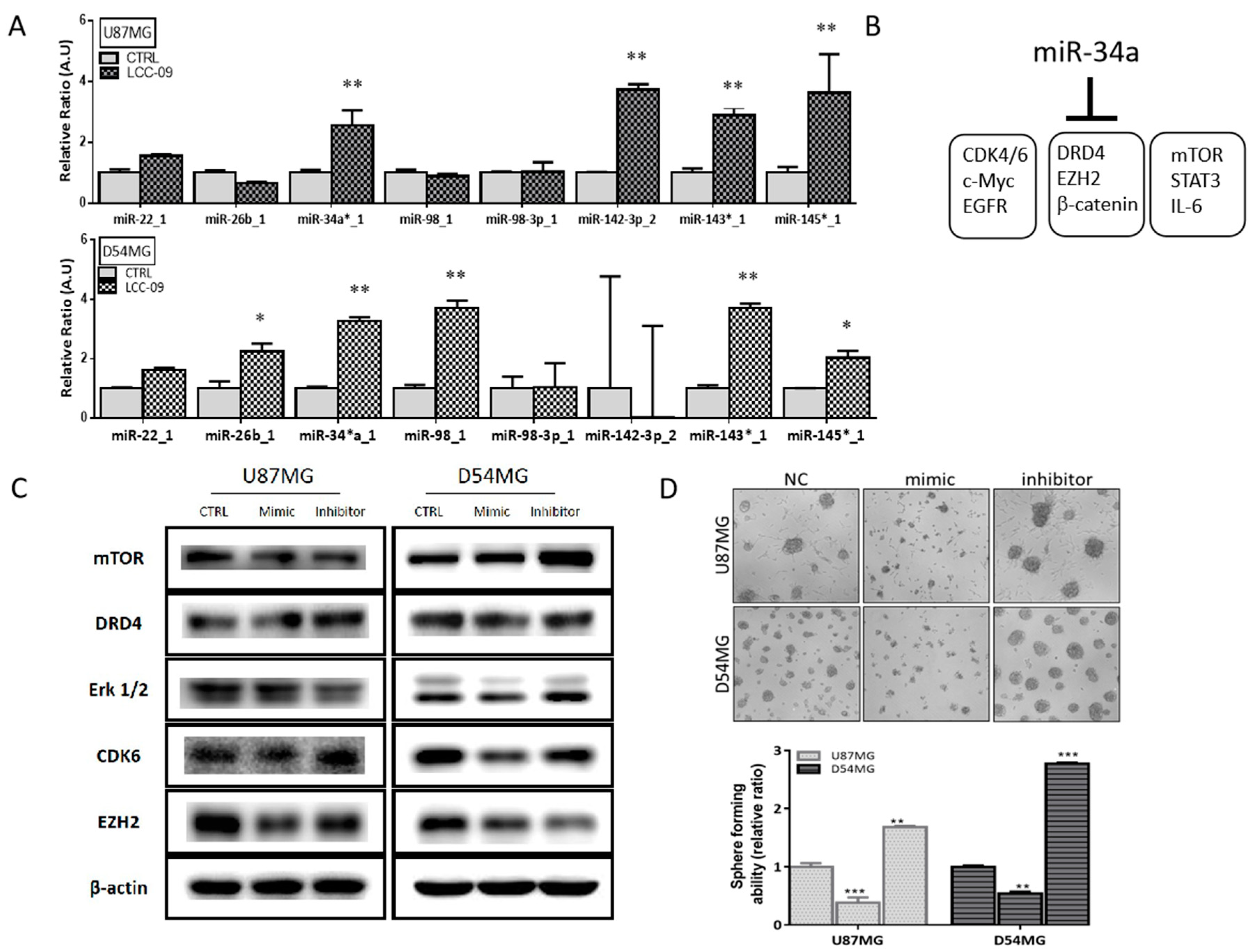
4.4. LCC-09, by Targeting Dopaminergic Signals and ALDH Activity, Induces Marked Attenuation of the Stem Cell-Like Phenotype of GBM Cells
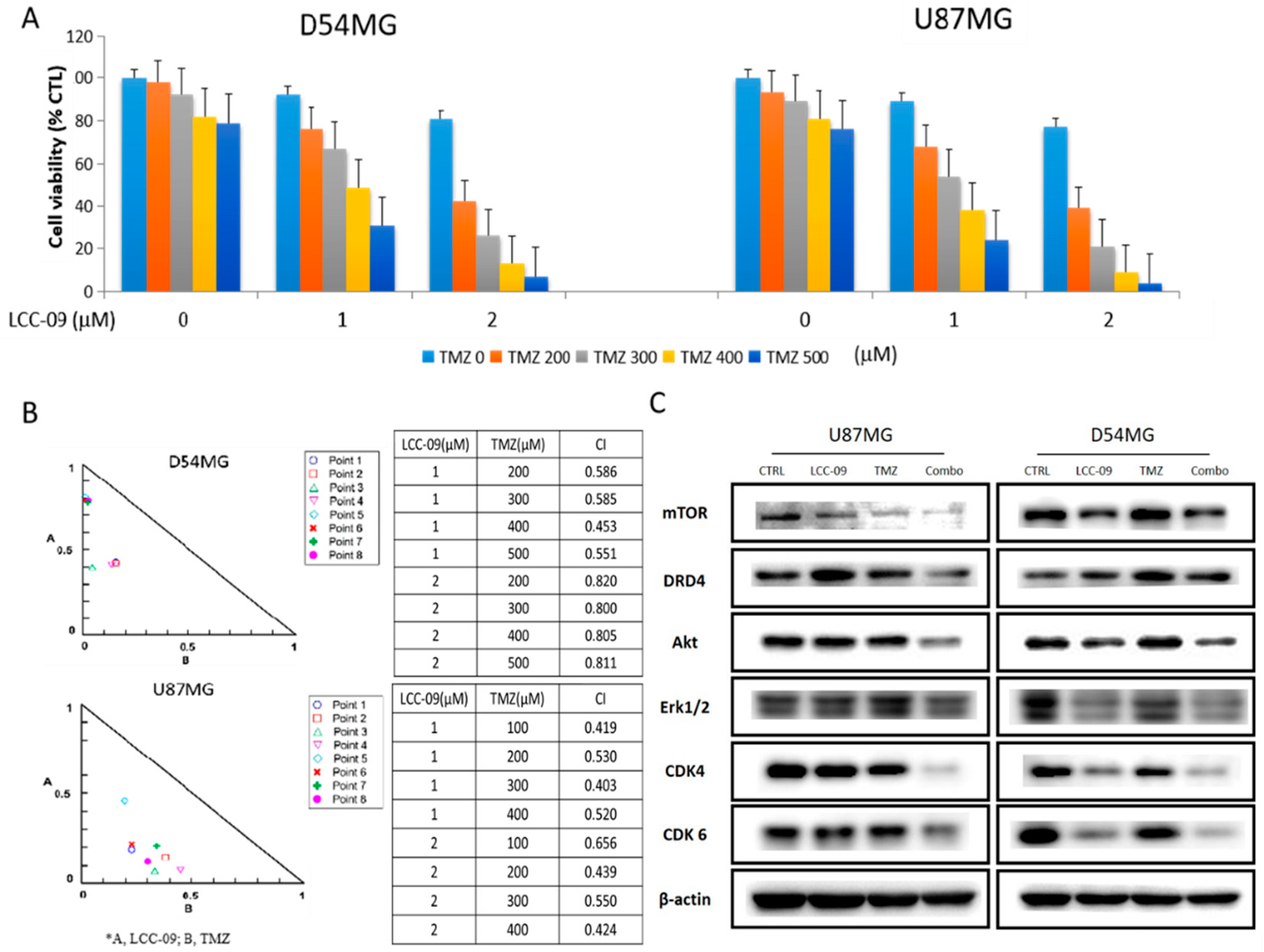
4.5. LCC-09 Synergistically Enhances the Anticancer Effect of TMZ in Therapy-Resistant GBM Cells
4.6. LCC-09 Suppresses GSC-Related Oncogenic and Dopaminergic Signals While Upregulating miR-34a in GBM Cells, In Vitro
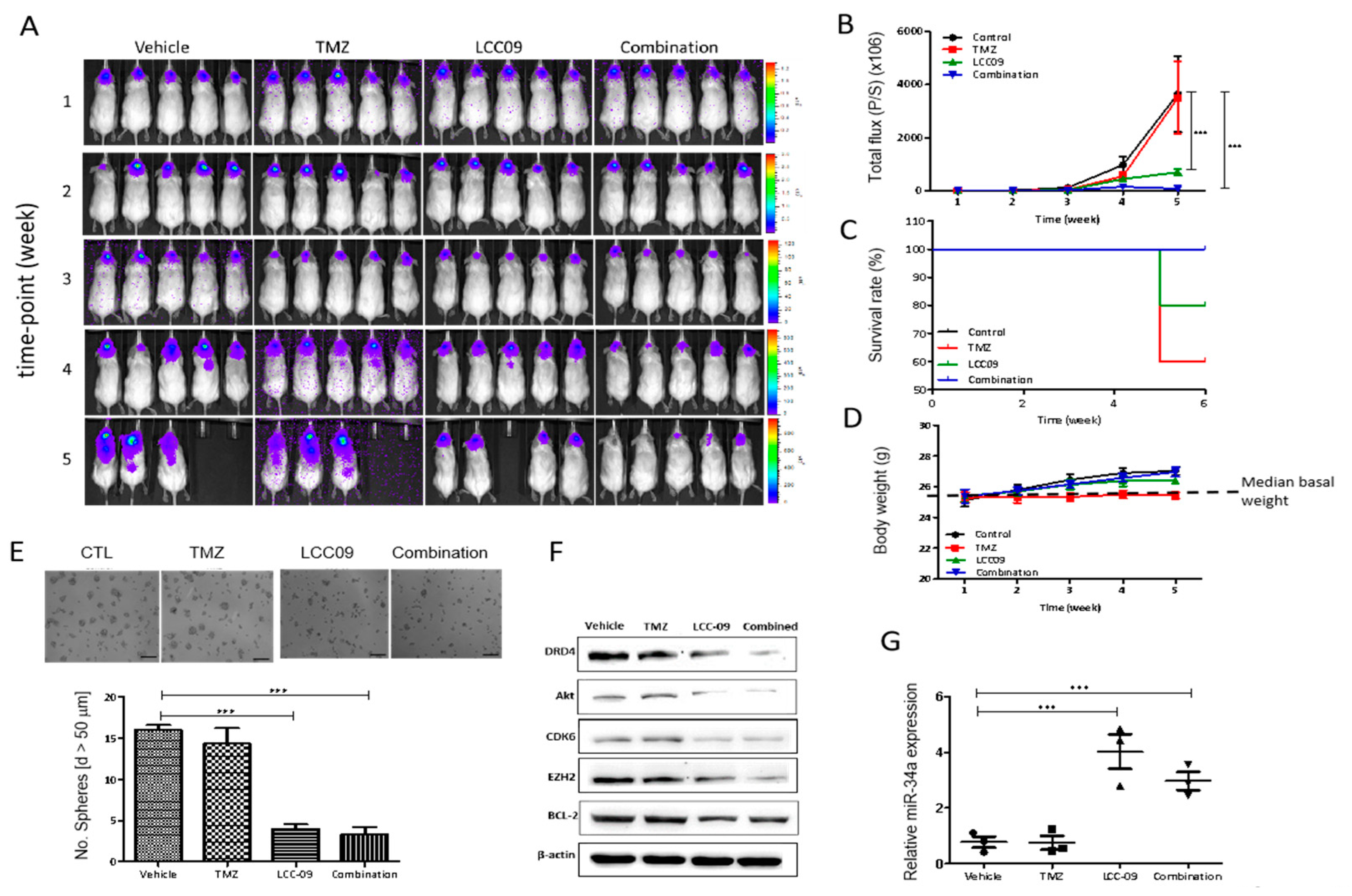
4.7. LCC-09 Significantly Suppresses GSC-Related Oncogenic and Dopaminergic Signals While Up-Regulating miR-34a in GBM Cells, In Vivo
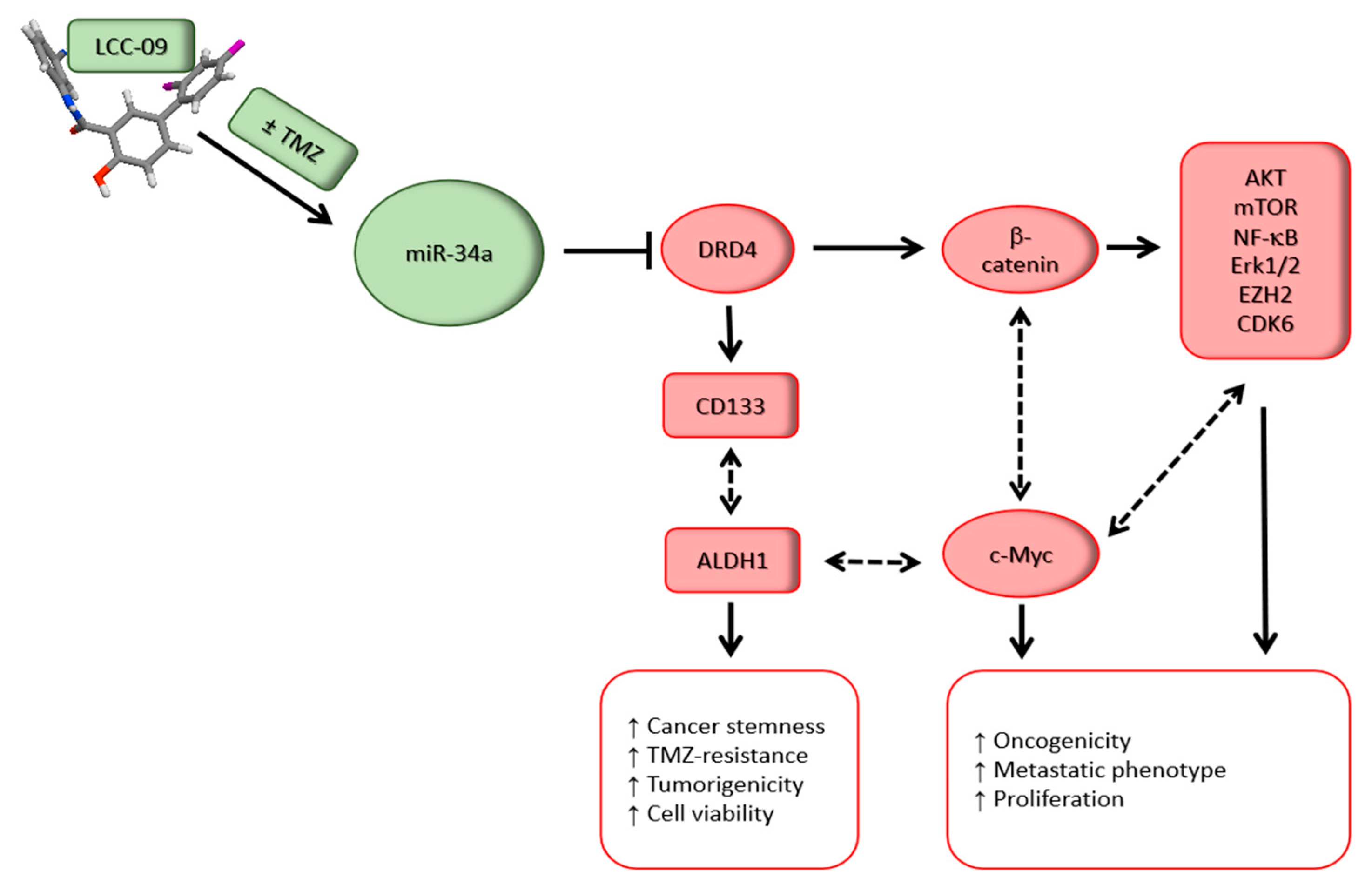
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.; Bankhead, A., 3rd; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.W.; Baldwin, A.S. IKK/nuclear factor-kappaB and oncogenesis: Roles in tumor-initiating cells and in the tumor microenvironment. Adv. Cancer Res. 2014, 121, 125–145. [Google Scholar]
- Kendellen, M.F.; Bradford, J.W.; Lawrence, C.L.; Clark, K.S.; Baldwin, A.S. Canonical and non-canonical NF-kappaB signaling promotes breast cancer tumor-initiating cells. Oncogene 2014, 33, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; Stifani, S. NF-κB Signalling in Glioblastoma. Biomedicines 2017, 5, 29. [Google Scholar] [CrossRef]
- Ferrandez, E.; Gutierrez, O.; Segundo, D.S.; Fernandez-Luna, J. NFκB activation in differentiating glioblastoma stem-like cells is promoted by hyaluronic acidsignaling through TLR4. Sci. Rep. 2018, 8, 6341. [Google Scholar] [CrossRef]
- Ohtsu, N.; Nakatani, Y.; Yamashita, D.; Ohue, S.; Ohnishi, T.; Kondo, T. Eva1 maintains the stem-like character of glioblastoma-initiating cells by activating the noncanonical NF-kappaB signaling pathway. Cancer Res. 2016, 76, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Yamini, B. NF-κB, Mesenchymal Differentiation and Glioblastoma. Cells 2018, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jin, X.; Sohn, Y.W.; Jeon, H.Y.; Kim, E.J.; Ham, S.W.; Jeon, H.M.; Chang, S.Y.; Oh, S.Y.; Yin, J.; et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett. 2014, 353, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Koch, L. Cancer: RANKL inhibition—A new weapon against breast cancer? Nat. Rev. Endocrinol. 2011, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Cronin, S.J.F.; Sigl, V.; Penninger, J.M. RANKL and RANK: From Mammalian Physiology to Cancer Treatment. Trends Cell Biol. 2018, 28, 213–223. [Google Scholar] [CrossRef]
- Lee, C.C.; Liu, F.L.; Chen, C.L.; Chen, T.C.; Chang, D.M.; Huang, H.S. Discovery of 5-(2′,4′-difluorophenyl)-salicylanilides as new inhibitors of receptor activator of NF-κB ligand (RANKL)-induced osteoclastogenesis. Eur. J. Med. Chem. 2015, 98, 115–126. [Google Scholar] [CrossRef]
- Dolma, S.; Selvadurai, H.J.; Lan, X.; Lee, L.; Kushida, M.; Voisin, V.; Whetstone, H.; So, M.; Aviv, T.; Park, N.; et al. Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell 2016, 29, 859–873. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, W.; Wang, F.; Wu, Q.; Li, W.; Zhong, X.; Tian, K.; Zeng, T.; Gao, L.; Liu, Y.; et al. Paired related homeobox 1 transactivates dopamine D2 receptor to maintain propagation and tumorigenicity of glioma-initiating cells. J. Mol. Cell Biol. 2017, 9, 302–314. [Google Scholar] [CrossRef]
- Ozawa, T.; James, C.D. Establishing intracranial brain tumor xenografts with subsequent analysis of tumor growth and response to therapy using bioluminescence imaging. J. Vis. Exp. 2010, 13, e1986. [Google Scholar] [CrossRef]
- Houghton, P.J.; Stewart, C.F.; Cheshire, P.J.; Richmond, L.B.; Kirstein, M.N.; Poquette, C.A.; Tan, M.; Friedman, H.S.; Brent, T.P. Antitumor activity of temozolomide combined with irinotecan is partly independent of O6-methylguanine-DNA methyltransferase and mismatch repair phenotypes in xenograft models. Clin. Cancer Res. 2000, 6, 4110–4118. [Google Scholar]
- Gfeller, D.; Michielin, O.; Zoete, V. Shaping the interaction landscape of bioactive molecules. Bioinformatics (Oxf. Engl.) 2013, 29, 3073–3079. [Google Scholar] [CrossRef]
- Osswald, M.; Blaes, J.; Liao, Y.; Solecki, G.; Gommel, M.; Berghoff, A.S.; Salphati, L.; Wallin, J.J.; Phillips, H.S.; Wick, W.; et al. Impact of Blood-Brain Barrier Integrity on Tumor Growth and Therapy Response in Brain Metastases. Clin. Cancer Res. 2016, 22, 6078–6087. [Google Scholar] [CrossRef]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef] [Green Version]
- Garnier, D.; Meehan, B.; Kislinger, T.; Daniel, P.; Sinha, A.; Abdulkarim, B.; Nakano, I.; Rak, J. Divergent evolution of temozolomide resistance in glioblastoma stem cells is reflected in extracellular vesicles and coupled with radiosensitization. Neuro Oncol. 2018, 20, 236–248. [Google Scholar] [CrossRef]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol. Med. Chir. (Tokyo) 2018, 58, 405–421. [Google Scholar] [CrossRef] [Green Version]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells - much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma Stem Cells Microenvironment: The Paracrine Roles of the Niche in Drug and Radioresistance. Stem Cells Int. 2016, 2016, 6809105. [Google Scholar] [CrossRef]
- Schäfer, A.; Teufel, J.; Ringel, F.; Bettstetter, M.; Hoepner, I.; Rasper, M.; Gempt, J.; Koeritzer, J.; Schmidt-Graf, F.; Meyer, B.; et al. Aldehyde dehydrogenase 1A1--a new mediator of resistance to temozolomide in glioblastoma. Neuro Oncol. 2012, 14, 1452–1464. [Google Scholar] [CrossRef]
- Takahashi, R.U.; Miyazaki, H.; Ochiya, T. The role of microRNAs in the regulation of cancer stem cells. Front. Genet. 2014, 4, 295. [Google Scholar] [CrossRef] [Green Version]
- Rathod, S.S.; Rani, S.B.; Khan, M.; Muzumdar, D.; Shiras, A. Tumor suppressive miRNA-34a suppresses cell proliferation and tumor growth of glioma stem cells by targeting Akt and Wnt signaling pathways. FEBS Open Bio 2014, 4, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Guessous, F.; Zhang, Y.; Dipierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jiang, J.; Yang, Y.; Schmittgen, T.D.; et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009, 69, 7569–7576. [Google Scholar] [CrossRef]
- Caragher, S.P.; Park, C.H.; Atashi, F.; Baisiwala, S.; Ahmed, A.U. Dopamine signaling and therapeutic resistance in GBM. Cancer Res. 2017, 77. [Google Scholar] [CrossRef]
- Caragher, S.P.; Hall, R.R.; Ahsan, R.; Ahmed, A.U. Monoamines in glioblastoma: Complex biology with therapeutic potential. Neuro Oncol. 2018, 20, 1014–1025. [Google Scholar] [CrossRef]
- Brescia, P.; Ortensi, B.; Fornasari, L.; Levi, D.; Broggi, G.; Pelicci, G. CD133 is essential for glioblastoma stem cell maintenance. Stem Cells 2013, 31, 857–869. [Google Scholar] [CrossRef]
- Liu, Q.; Nguyen, D.H.; Dong, Q.; Shitaku, P.; Chung, K.; Liu, O.Y.; Tso, J.L.; Liu, J.Y.; Konkankit, V.; Cloughesy, T.F.; et al. Molecular properties of CD133+ glioblastoma stem cells derived from treatment-refractory recurrent brain tumors. J. Neurooncol. 2009, 94, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Kuol, N.; Stojanovska, L.; Apostolopoulos, V.; Nurgali, K. Role of the nervous system in cancer metastasis. J. Exp. Clin. Cancer Res. 2018, 37, 5. [Google Scholar] [CrossRef]
- Doorn, J.A.; Florang, V.R.; Schamp, J.H.; Vanle, B.C. Aldehyde dehydrogenase inhibition generates a reactive dopamine metabolite autotoxic to dopamine neurons. Parkinsonism Relat. Disord. 2014, 20 (Suppl. 1), S73–S75. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Moller, H.G.; Rasmussen, A.P.; Andersen, H.H.; Johnsen, K.B.; Henriksen, M.; Duroux, M. A systematic review of microRNA in glioblastoma multiforme: Micro-modulators in the mesenchymal mode of migration and invasion. Mol. Neurobiol. 2013, 47, 131–144. [Google Scholar] [CrossRef]
- Ofek, P.; Calderon, M.; Mehrabadi, F.S.; Krivitsky, A.; Ferber, S.; Tiram, G.; Yerushalmi, N.; Kredo-Russo, S.; Grossman, R.; Ram, Z.; et al. Restoring the oncosuppressor activity of microRNA-34a in glioblastoma using a polyglycerol-based polyplex. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 2201–2214. [Google Scholar] [CrossRef] [Green Version]
- Ahir, B.K.; Ozer, H.; Engelhard, H.H.; Lakka, S.S. MicroRNAs in glioblastoma pathogenesis and therapy: A comprehensive review. Crit. Rev. Oncol. Hematol. 2017, 120, 22–33. [Google Scholar] [CrossRef]
- Parodi, F.; Carosio, R.; Ragusa, M.; Di Pietro, C.; Maugeri, M.; Barbagallo, D.; Sallustio, F.; Allemanni, G.; Pistillo, M.P.; Casciano, I.; et al. Epigenetic dysregulation in neuroblastoma: A tale of miRNAs and DNA methylation. Biochim. Biophys. Acta 2016, 1859, 1502–1514. [Google Scholar] [CrossRef]
- Bader, A.G. miR-34—A microRNA replacement therapy is headed to the clinic. Front. Genet. 2012, 3, 120. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Andreieva, S.V.; Korets, K.V.; Mykytenko, D.O.; Baklaushev, V.P.; Huleyuk, N.L.; Kovalova, O.A.; Kotsarenko, K.V.; Chekhonin, V.P.; Vassetzky, Y.S.; et al. Temozolomide promotes genomic and phenotypic changes in glioblastoma cells. Cancer Cell Int. 2016, 16, 36. [Google Scholar] [CrossRef]
- Berg, K.A.; Clarke, W.P. Making Sense of Pharmacology: Inverse Agonism and Functional Selectivity. Int. J. Neuropsychopharmacol. 2018, 21, 962–977. [Google Scholar] [CrossRef] [Green Version]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, Y.-T.; Wu, A.T.; Bamodu, O.A.; Wei, L.; Lin, C.-M.; Yen, Y.; Chao, T.-Y.; Mukhopadhyay, D.; Hsiao, M.; Huang, H.-S. A Novel Multi-Target Small Molecule, LCC-09, Inhibits Stemness and Therapy-Resistant Phenotypes of Glioblastoma Cells by Increasing miR-34a and Deregulating the DRD4/Akt/mTOR Signaling Axis. Cancers 2019, 11, 1442. https://doi.org/10.3390/cancers11101442
Wen Y-T, Wu AT, Bamodu OA, Wei L, Lin C-M, Yen Y, Chao T-Y, Mukhopadhyay D, Hsiao M, Huang H-S. A Novel Multi-Target Small Molecule, LCC-09, Inhibits Stemness and Therapy-Resistant Phenotypes of Glioblastoma Cells by Increasing miR-34a and Deregulating the DRD4/Akt/mTOR Signaling Axis. Cancers. 2019; 11(10):1442. https://doi.org/10.3390/cancers11101442
Chicago/Turabian StyleWen, Ya-Ting, Alexander TH Wu, Oluwaseun Adebayo Bamodu, Li Wei, Chien-Min Lin, Yun Yen, Tsu-Yi Chao, Debabrata Mukhopadhyay, Michael Hsiao, and Hsu-Shan Huang. 2019. "A Novel Multi-Target Small Molecule, LCC-09, Inhibits Stemness and Therapy-Resistant Phenotypes of Glioblastoma Cells by Increasing miR-34a and Deregulating the DRD4/Akt/mTOR Signaling Axis" Cancers 11, no. 10: 1442. https://doi.org/10.3390/cancers11101442
APA StyleWen, Y. -T., Wu, A. T., Bamodu, O. A., Wei, L., Lin, C. -M., Yen, Y., Chao, T. -Y., Mukhopadhyay, D., Hsiao, M., & Huang, H. -S. (2019). A Novel Multi-Target Small Molecule, LCC-09, Inhibits Stemness and Therapy-Resistant Phenotypes of Glioblastoma Cells by Increasing miR-34a and Deregulating the DRD4/Akt/mTOR Signaling Axis. Cancers, 11(10), 1442. https://doi.org/10.3390/cancers11101442