STAT3 Activity Promotes Programmed-Death Ligand 1 Expression and Suppresses Immune Responses in Breast Cancer

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
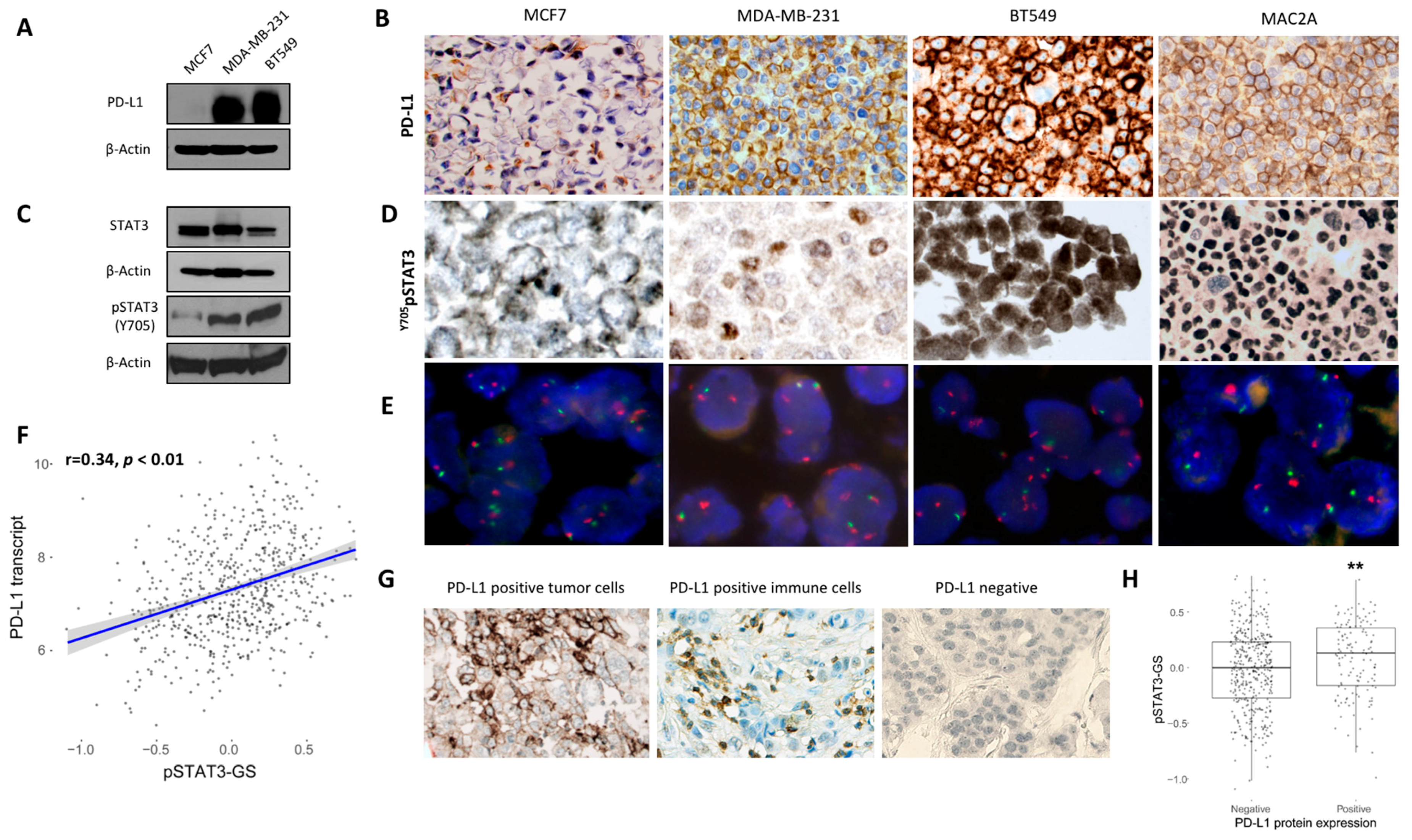
2.1. Association Between PD-L1 and STAT3 Expression in Breast Cancer Cell Lines and Human Breast Cancer
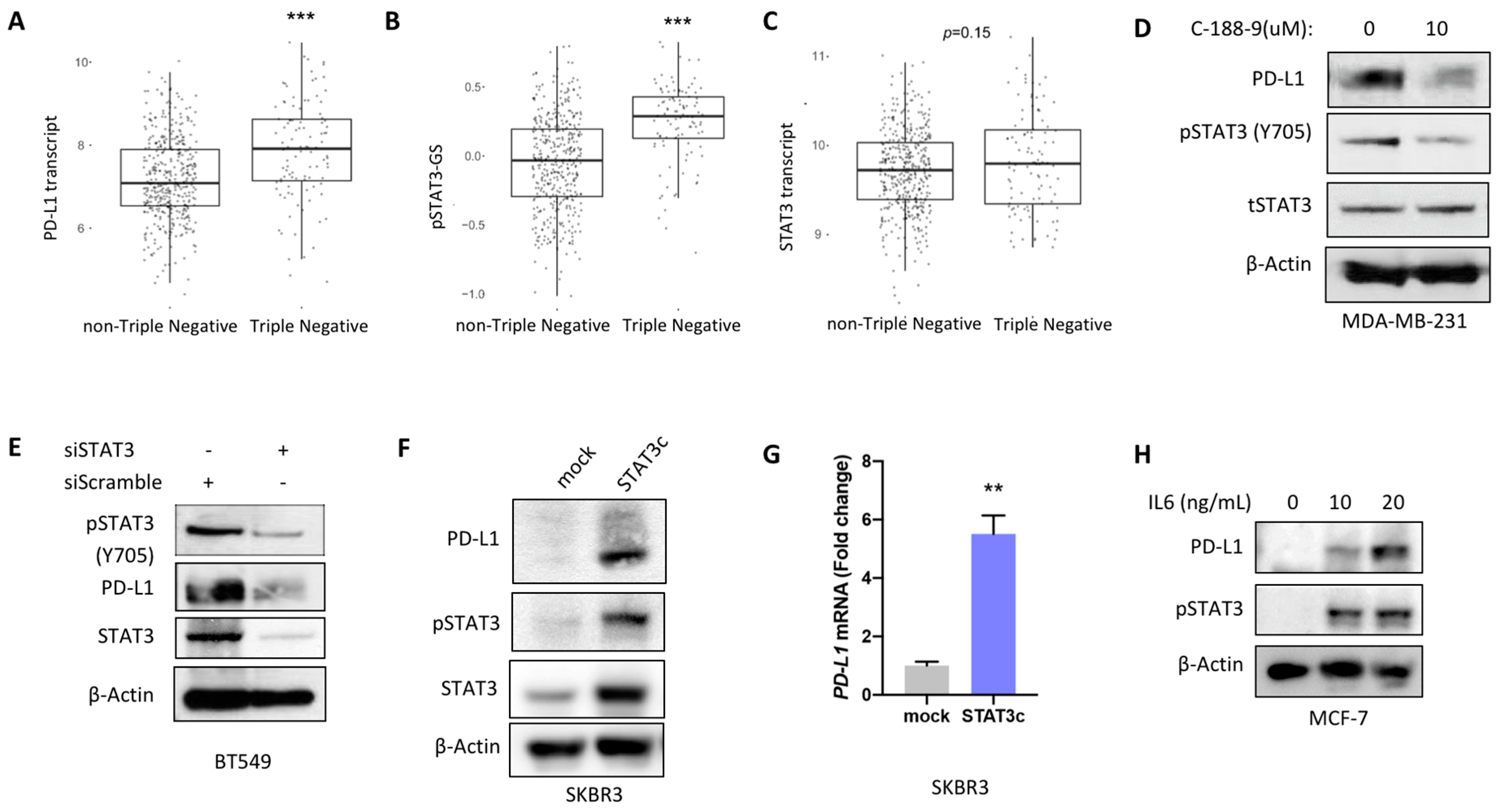
2.2. STAT3 Mediated Regulation of PD-L1 Expression In Vitro
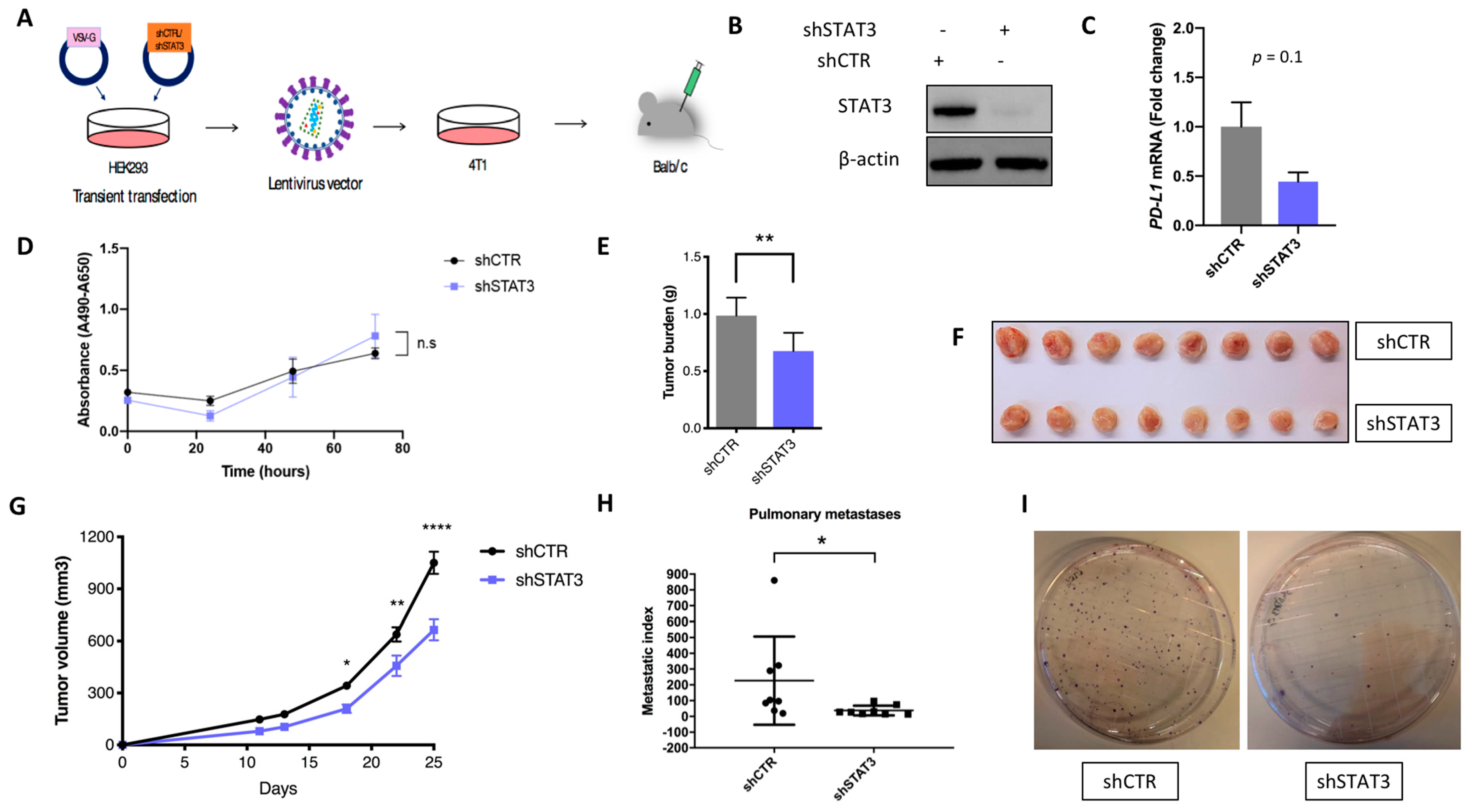
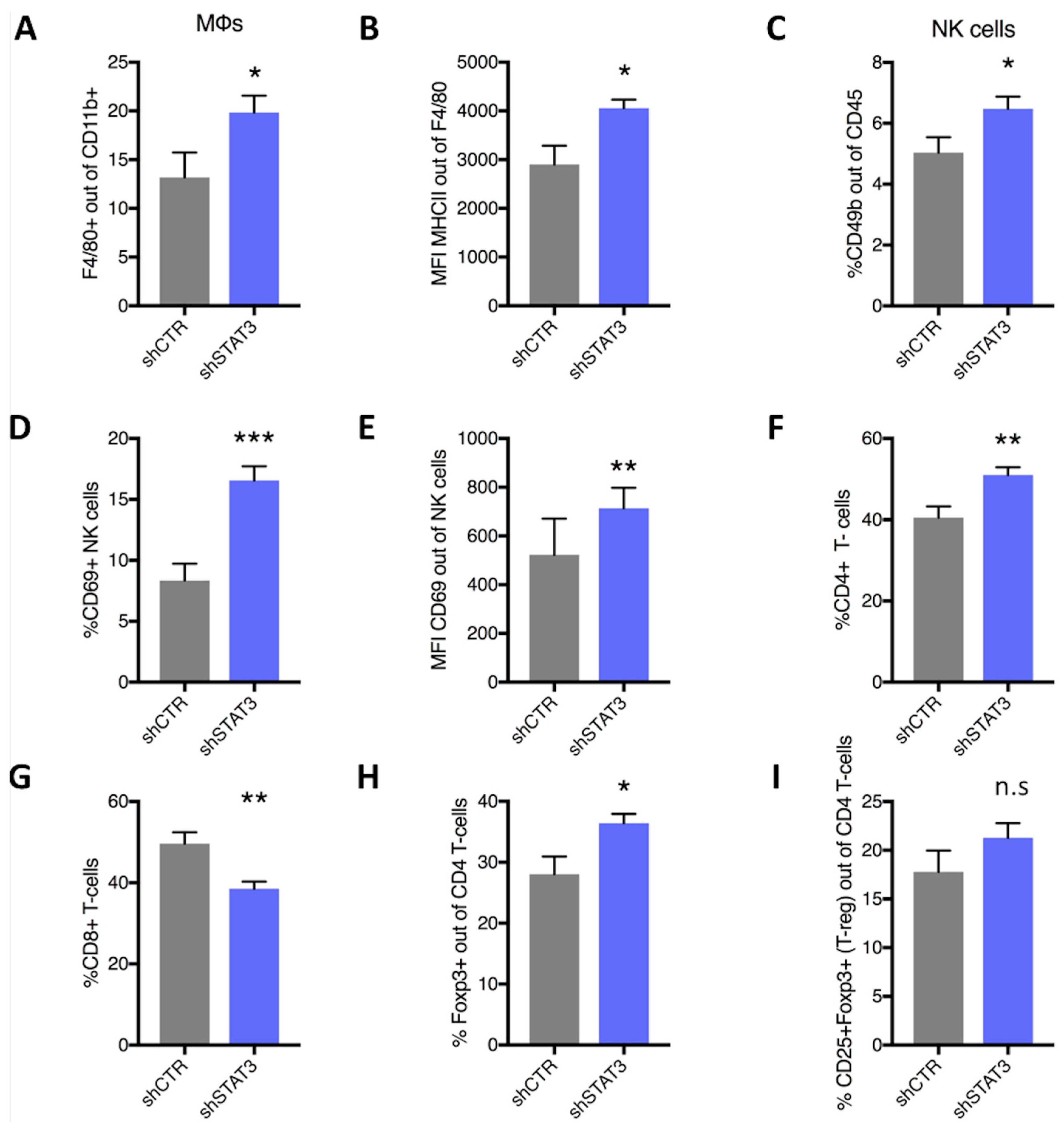
2.3. Stat3 Silencing Downregulates PD-L1 in Mouse Cells, Restricts Tumor Growth and Metastatic Formation and Modifies Anti-Tumor Immune Response In Vivo
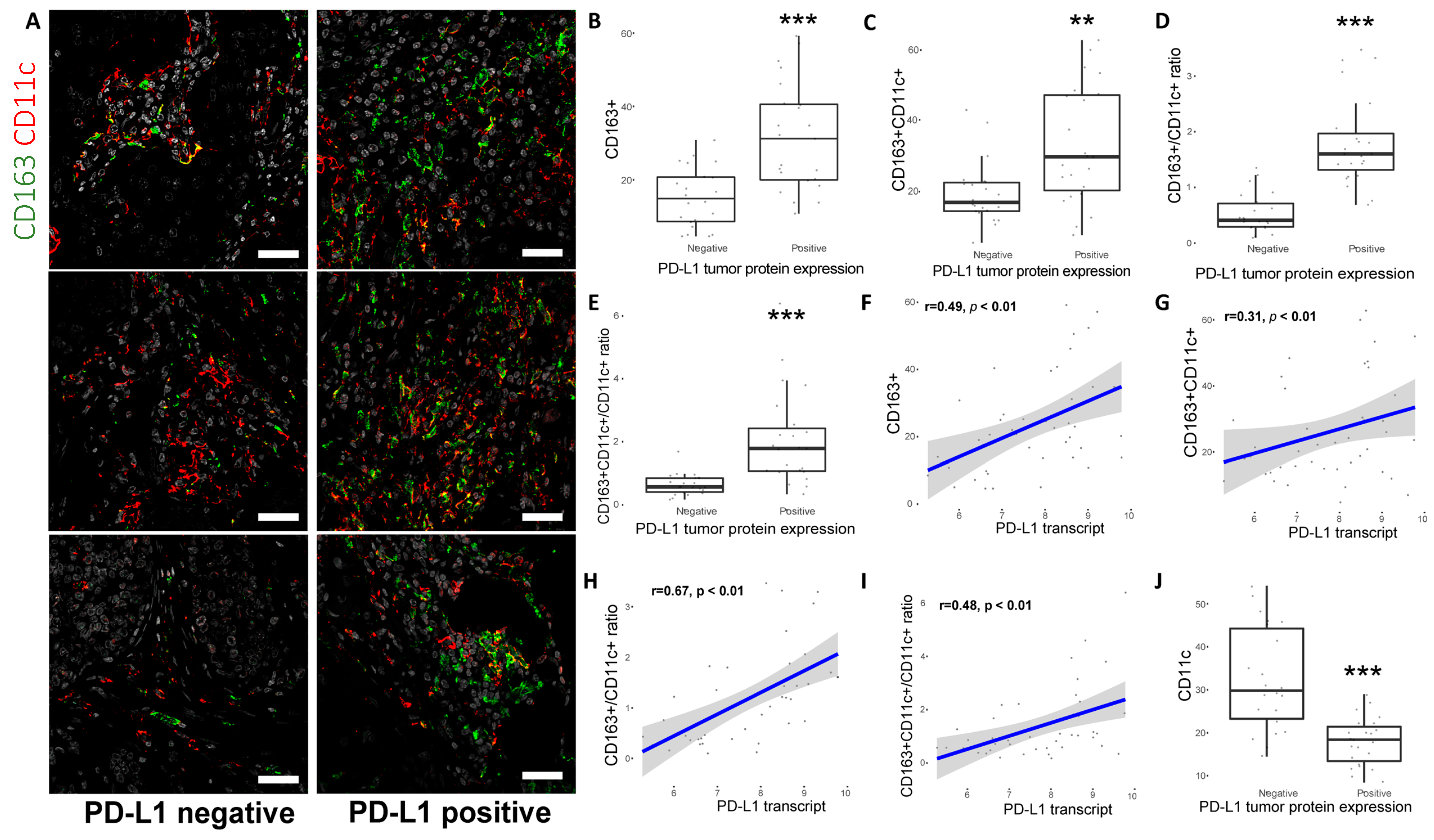
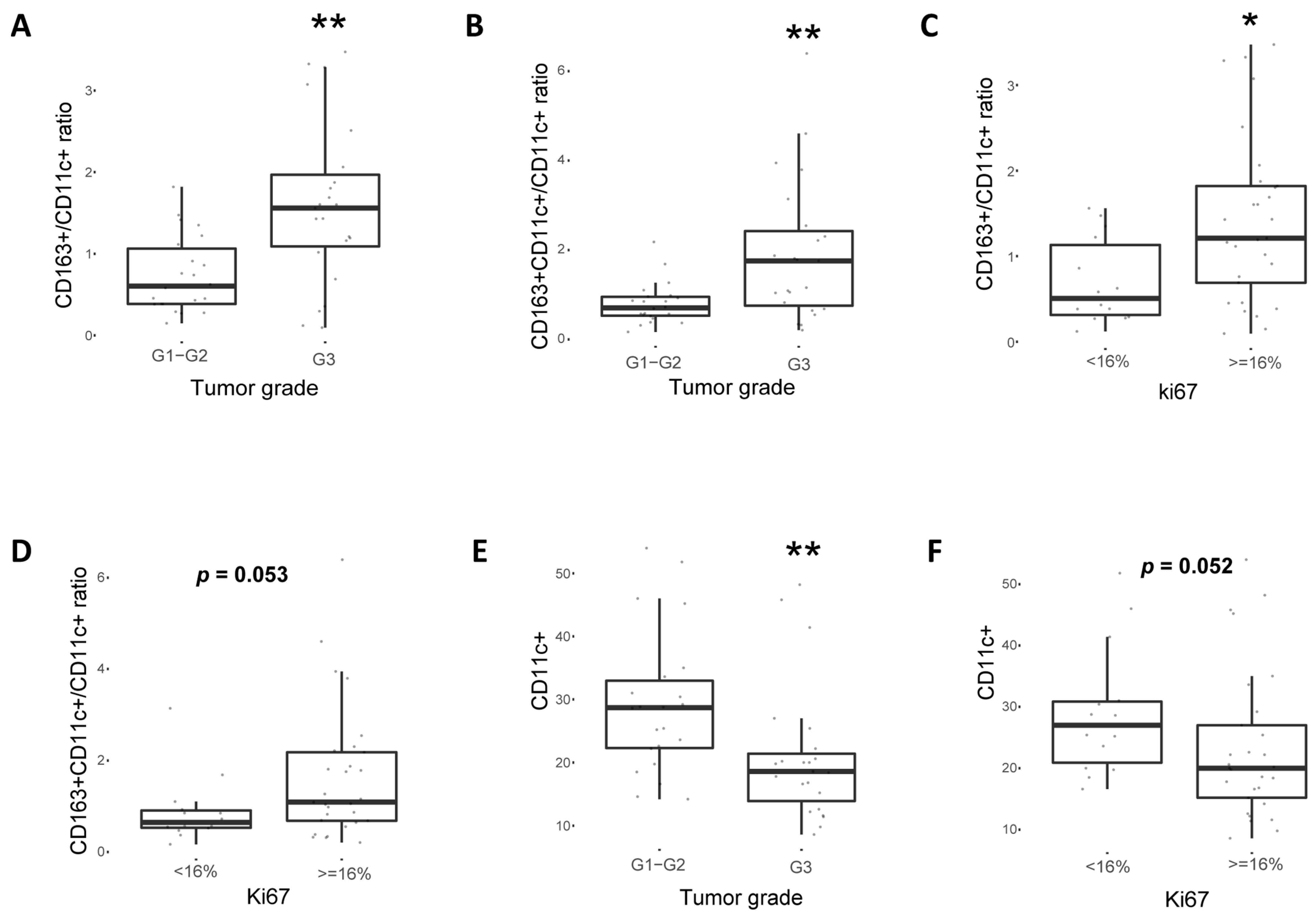
2.4. CD163+ TAM Phenotype Is Associated with Higher PD-L1 Expression, Grade and Proliferation in Breast Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Plasmids, and Reagents
4.2. Western Blot Analysis
4.3. RNA Extraction, cDNA Synthesis, and Real Time Quantitative Polymerase Chain Reaction (RT-qPCR)
4.4. Tissue Microarrays, Immunohistochemical Methods and Scoring
4.5. Fluorescence in Situ Hybridization
4.6. Transient Transfections and Gene Silencing
4.7. Proliferation Assay
4.8. Lentiviral Vectors
4.9. 4T1 Breast Cancer Animal Model and Tumor Growth Assessment
4.10. Tumor Dissociation
4.11. Flow Cytometry
4.12. Lung Metastasis Colony Assay
4.13. Immunofluorescence for Macrophage Markers in Human Breast Tumors
4.14. Patient Cohorts
4.15. Gene Expression Profiling Using Microarrays
4.16. Preprocessing and Normalization of the Microarray Gene Expression Data
4.17. Phosphorylated STAT3-Associated Gene Signature
4.18. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Carbognin, L.; De Silva, P.; Criscitiello, C.; Lambertini, M. Tumor-infiltrating lymphocytes in breast cancer according to tumor subtype: Current state of the art. Breast 2017, 35, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Gombos, A.; Latifyan, S.; Piccart-Gebhart, M.; Kok, M.; Buisseret, L. Targeting immune checkpoints in breast cancer: An update of early results. ESMO Open 2017, 2, e000255. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Zerdes, I.; Matikas, A.; Bergh, J.; Rassidakis, G.Z.; Foukakis, T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: Biology and clinical correlations. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsaves, V.; Tsesmetzis, N.; Chioureas, D.; Kis, L.; Leventaki, V.; Drakos, E.; Panaretakis, T.; Grander, D.; Medeiros, L.J.; Young, K.H.; et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia 2017, 31, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—Using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Wallerius, M.; Wallmann, T.; Bartish, M.; Ostling, J.; Mezheyeuski, A.; Tobin, N.P.; Nygren, E.; Pangigadde, P.; Pellegrini, P.; Squadrito, M.L.; et al. Guidance Molecule SEMA3A Restricts Tumor Growth by Differentially Regulating the Proliferation of Tumor-Associated Macrophages. Cancer Res. 2016, 76, 3166–3178. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef]
- Frank, D.A. Transcription factor STAT3 as a prognostic marker and therapeutic target in cancer. J. Clin. Oncol. 2013, 31, 4560–4561. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Toor, S.M.; Ali, B.R.; Elkord, E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin. Ther. Targets 2018, 22, 547–557. [Google Scholar] [CrossRef]
- Zhang, N.; Zeng, Y.; Du, W.; Zhu, J.; Shen, D.; Liu, Z.; Huang, J.A. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int. J. Oncol. 2016, 49, 1360–1368. [Google Scholar] [CrossRef] [Green Version]
- Sumimoto, H.; Takano, A.; Teramoto, K.; Daigo, Y. RAS-Mitogen-Activated Protein Kinase Signal Is Required for Enhanced PD-L1 Expression in Human Lung Cancers. PLoS ONE 2016, 11, e0166626. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.L.; Yu, G.T.; Wu, L.; Mao, L.; Deng, W.W.; Liu, J.F.; Kulkarni, A.B.; Zhang, W.F.; Zhang, L.; Sun, Z.J. STAT3 Induces Immunosuppression by Upregulating PD-1/PD-L1 in HNSCC. J. Dent. Res. 2017, 96, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, J.; Etemadi, N.; Hollande, F.; Ernst, M.; Buchert, M. The JAK/STAT3 axis: A comprehensive drug target for solid malignancies. Semin. Cancer Biol. 2017, 45, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; Broz, M.L.; Ranger, J.J.; Ozcelik, J.; Ahn, R.; Zuo, D.; Ursini-Siegel, J.; Hallett, M.T.; Krummel, M.; Muller, W.J. STAT3 Establishes an Immunosuppressive Microenvironment during the Early Stages of Breast Carcinogenesis to Promote Tumor Growth and Metastasis. Cancer Res. 2016, 76, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Hartley, G.P.; Chow, L.; Ammons, D.T.; Wheat, W.H.; Dow, S.W. Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol. Res. 2018, 6, 1260–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. npj Breast Cancer 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Garlanda, C.; Jaillon, S.; Marone, G.; Mantovani, A. Tumor associated macrophages and neutrophils in tumor progression. J. Cell. Physiol. 2013, 228, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Dong, X.; Estrella, J.S.; Correa, A.M.; Xu, Y.; Hofstetter, W.L.; Sudo, K.; Onodera, H.; Suzuki, K.; Suzuki, A.; et al. Tumor-associated macrophage infiltration is highly associated with PD-L1 expression in gastric adenocarcinoma. Gastric Cancer 2018, 21, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Dhupkar, P.; Gordon, N.; Stewart, J.; Kleinerman, E.S. Anti-PD-1 therapy redirects macrophages from an M2 to an M1 phenotype inducing regression of OS lung metastases. Cancer Med. 2018, 7, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef]
- Liu, H.; Shen, J.; Lu, K. IL-6 and PD-L1 blockade combination inhibits hepatocellular carcinoma cancer development in mouse model. Biochem. Biophys. Res. Commun. 2017, 486, 239–244. [Google Scholar] [CrossRef]
- Lu, C.; Talukder, A.; Savage, N.M.; Singh, N.; Liu, K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology 2017, 6, e1291106. [Google Scholar] [CrossRef] [Green Version]
- Santoni, M.; Romagnoli, E.; Saladino, T.; Foghini, L.; Guarino, S.; Capponi, M.; Giannini, M.; Cognigni, P.D.; Ferrara, G.; Battelli, N. Triple negative breast cancer: Key role of Tumor-Associated Macrophages in regulating the activity of anti-PD-1/PD-L1 agents. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 78–84. [Google Scholar] [CrossRef]
- Atsaves, V.; Lekakis, L.; Drakos, E.; Leventaki, V.; Ghaderi, M.; Baltatzis, G.E.; Chioureas, D.; Jones, D.; Feretzaki, M.; Liakou, C.; et al. The oncogenic JUNB/CD30 axis contributes to cell cycle deregulation in ALK+ anaplastic large cell lymphoma. Br. J. Haematol. 2014, 167, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Cotta, C.V.; Leventaki, V.; Atsaves, V.; Vidaki, A.; Schlette, E.; Jones, D.; Medeiros, L.J.; Rassidakis, G.Z. The helix-loop-helix protein Id2 is expressed differentially and induced by myc in T-cell lymphomas. Cancer 2008, 112, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.; Lindstrom, L.S.; Harrell, J.C.; Falato, C.; Carlson, J.W.; Wright, P.K.; Foukakis, T.; Perou, C.M.; Czene, K.; Bergh, J.; et al. Gene Expression Signatures and Immunohistochemical Subtypes Add Prognostic Value to Each Other in Breast Cancer Cohorts. Clin. Cancer Res. 2017, 23, 7512–7520. [Google Scholar] [CrossRef] [PubMed]
- Follenzi, A.; Naldini, L. HIV-based vectors. Preparation and use. Methods Mol. Med. 2002, 69, 259–274. [Google Scholar] [PubMed]
- Witt, K.; Ligtenberg, M.A.; Conti, L.; Lanzardo, S.; Ruiu, R.; Wallmann, T.; Tufvesson-Stiller, H.; Chambers, B.J.; Rolny, C.; Lladser, A.; et al. Cripto-1 Plasmid DNA Vaccination Targets Metastasis and Cancer Stem Cells in Murine Mammary Carcinoma. Cancer Immunol. Res. 2018, 6, 1417–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casazza, A.; Fu, X.; Johansson, I.; Capparuccia, L.; Andersson, F.; Giustacchini, A.; Squadrito, M.L.; Venneri, M.A.; Mazzone, M.; Larsson, E.; et al. Systemic and targeted delivery of semaphorin 3A inhibits tumor angiogenesis and progression in mouse tumor models. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef]
- Bengtsson, H.; Simpson, K.; Bullard, J.; Hansen, K. Aroma. Affymetrix: A Generic Framework in R for Analyzing Small to Very Large Affymetrix Data Sets in Bounded Memory; Tech Report; University of California, Berkeley: Berkeley, CA, USA, 2008. [Google Scholar]
- Sonnenblick, A.; Brohee, S.; Fumagalli, D.; Vincent, D.; Venet, D.; Ignatiadis, M.; Salgado, R.; Van den Eynden, G.; Rothe, F.; Desmedt, C.; et al. Constitutive phosphorylated STAT3-associated gene signature is predictive for trastuzumab resistance in primary HER2-positive breast cancer. BMC Med. 2015, 13, 177. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID gene ID conversion tool. Bioinformation 2008, 2, 428–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendoo, D.M.; Ratanasirigulchai, N.; Schröder, M.S.; Paré, L.; Parker, J.S.; Prat, A.; Haibe-Kains, B. Genefu: An R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 2015, 32, 1097–1099. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zerdes, I.; Wallerius, M.; Sifakis, E.G.; Wallmann, T.; Betts, S.; Bartish, M.; Tsesmetzis, N.; Tobin, N.P.; Coucoravas, C.; Bergh, J.; et al. STAT3 Activity Promotes Programmed-Death Ligand 1 Expression and Suppresses Immune Responses in Breast Cancer. Cancers 2019, 11, 1479. https://doi.org/10.3390/cancers11101479
Zerdes I, Wallerius M, Sifakis EG, Wallmann T, Betts S, Bartish M, Tsesmetzis N, Tobin NP, Coucoravas C, Bergh J, et al. STAT3 Activity Promotes Programmed-Death Ligand 1 Expression and Suppresses Immune Responses in Breast Cancer. Cancers. 2019; 11(10):1479. https://doi.org/10.3390/cancers11101479
Chicago/Turabian StyleZerdes, Ioannis, Majken Wallerius, Emmanouil G. Sifakis, Tatjana Wallmann, Stina Betts, Margarita Bartish, Nikolaos Tsesmetzis, Nicholas P. Tobin, Christos Coucoravas, Jonas Bergh, and et al. 2019. "STAT3 Activity Promotes Programmed-Death Ligand 1 Expression and Suppresses Immune Responses in Breast Cancer" Cancers 11, no. 10: 1479. https://doi.org/10.3390/cancers11101479
APA StyleZerdes, I., Wallerius, M., Sifakis, E. G., Wallmann, T., Betts, S., Bartish, M., Tsesmetzis, N., Tobin, N. P., Coucoravas, C., Bergh, J., Rassidakis, G. Z., Rolny, C., & Foukakis, T. (2019). STAT3 Activity Promotes Programmed-Death Ligand 1 Expression and Suppresses Immune Responses in Breast Cancer. Cancers, 11(10), 1479. https://doi.org/10.3390/cancers11101479