Simultaneous Inhibition of BCR-ABL1 Tyrosine Kinase and PAK1/2 Serine/Threonine Kinase Exerts Synergistic Effect against Chronic Myeloid Leukemia Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
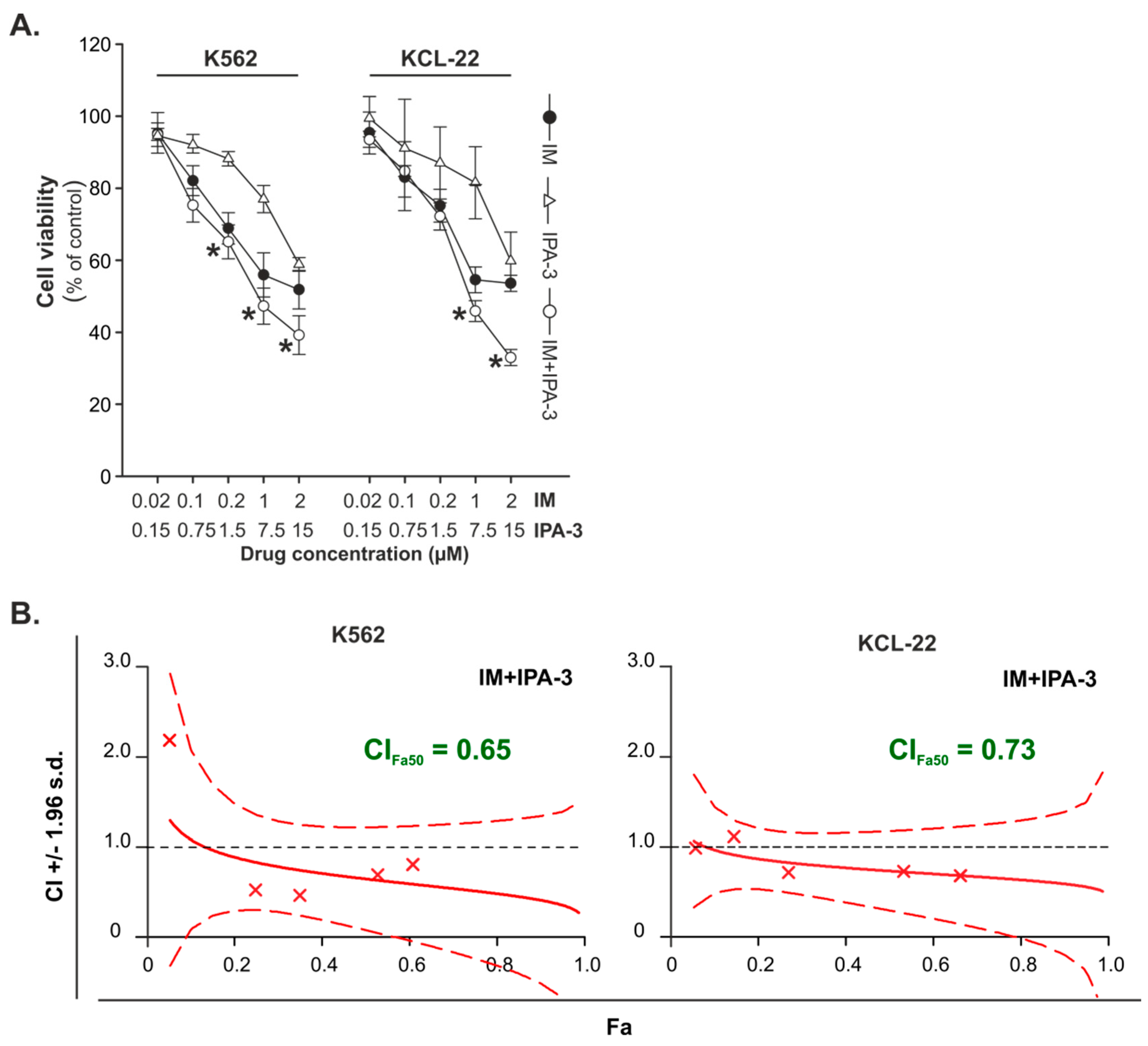
2.1. Effects of Combination Treatment of IM with IPA-3 against CML-BP Cell Lines
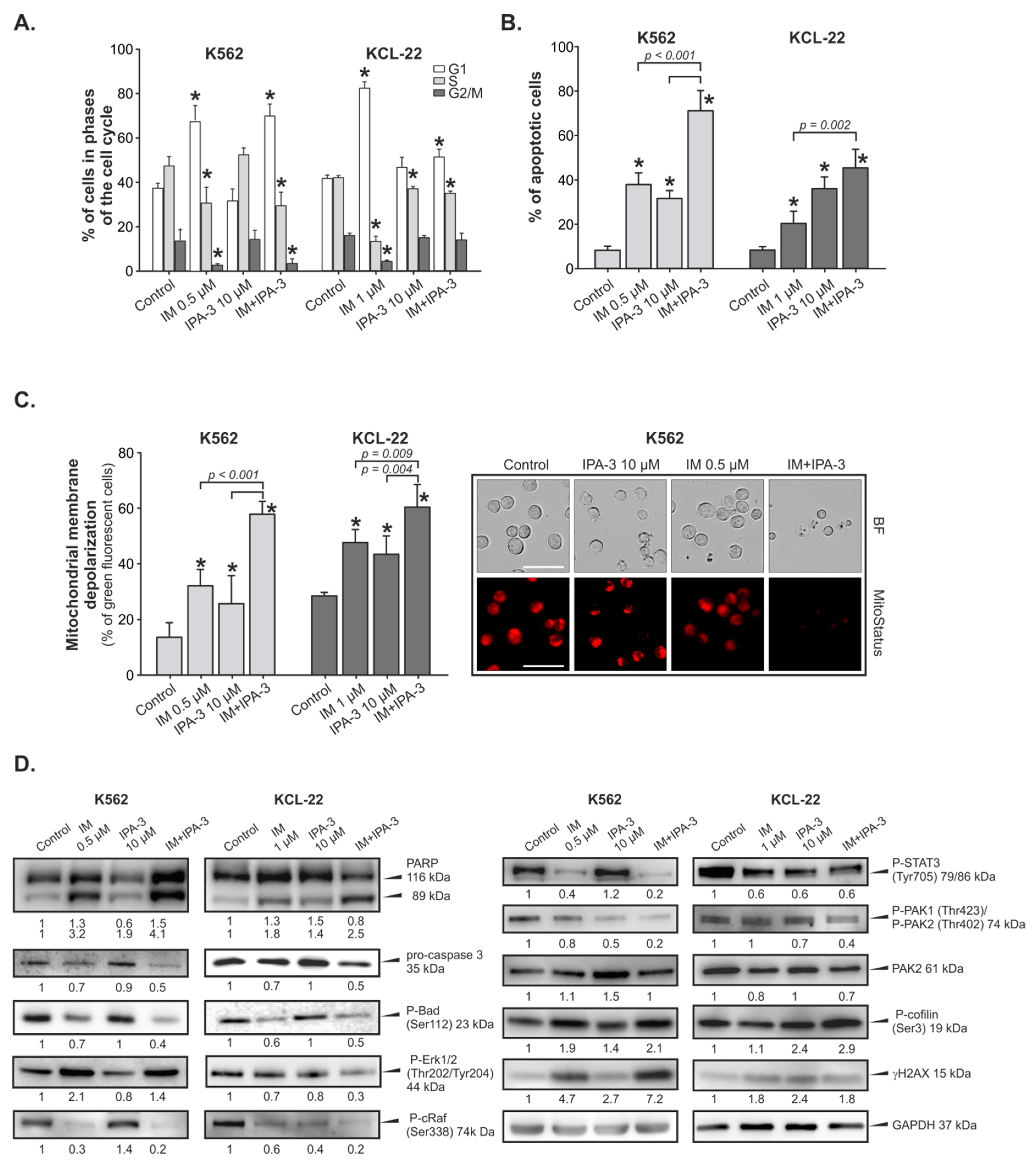
2.2. Cell Cycle Perturbations and Induction of Apoptosis by IM with/without IPA-3
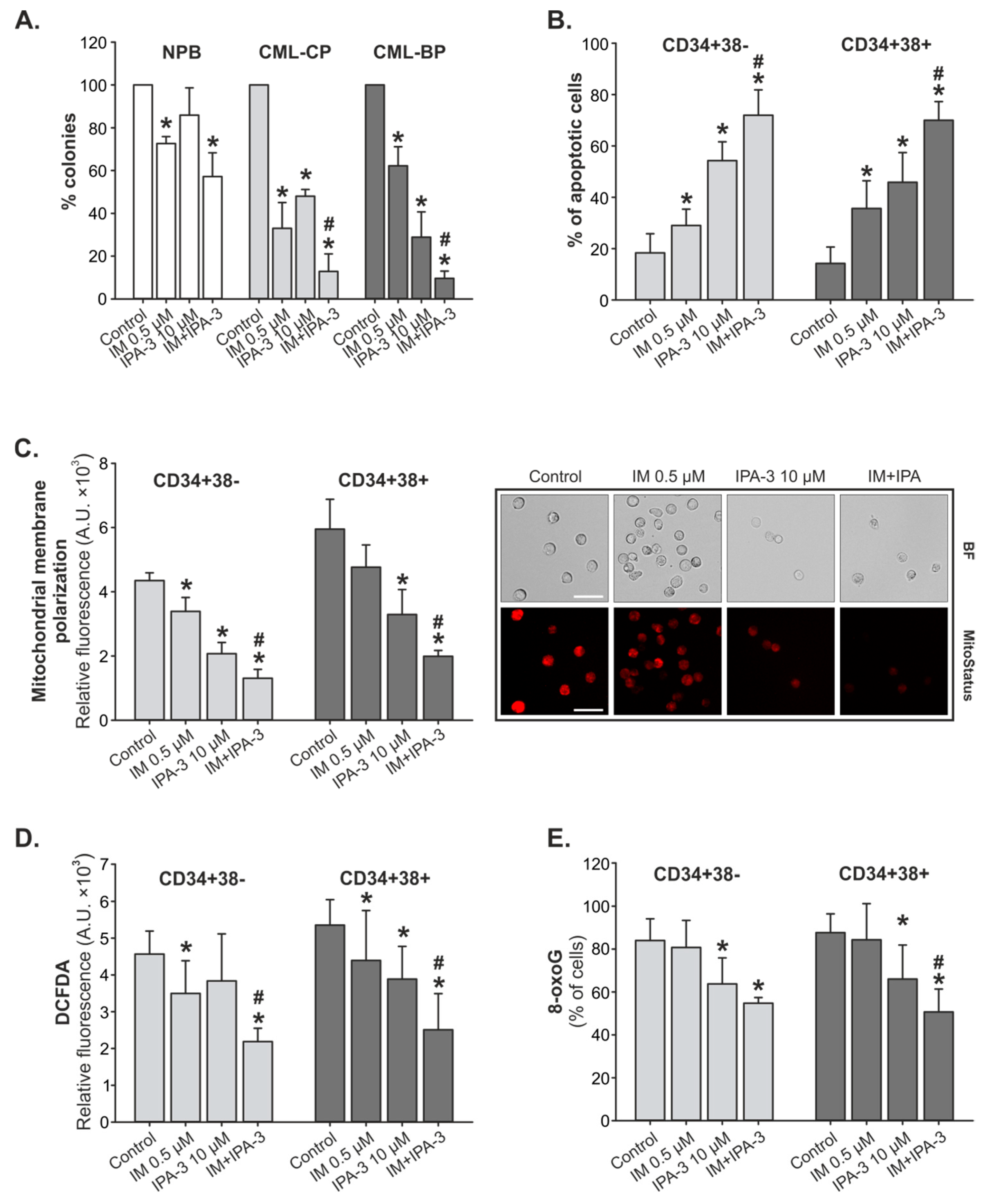
2.3. Response of Primary CML-CP and CML-BP Cells to IM with/without IPA-3
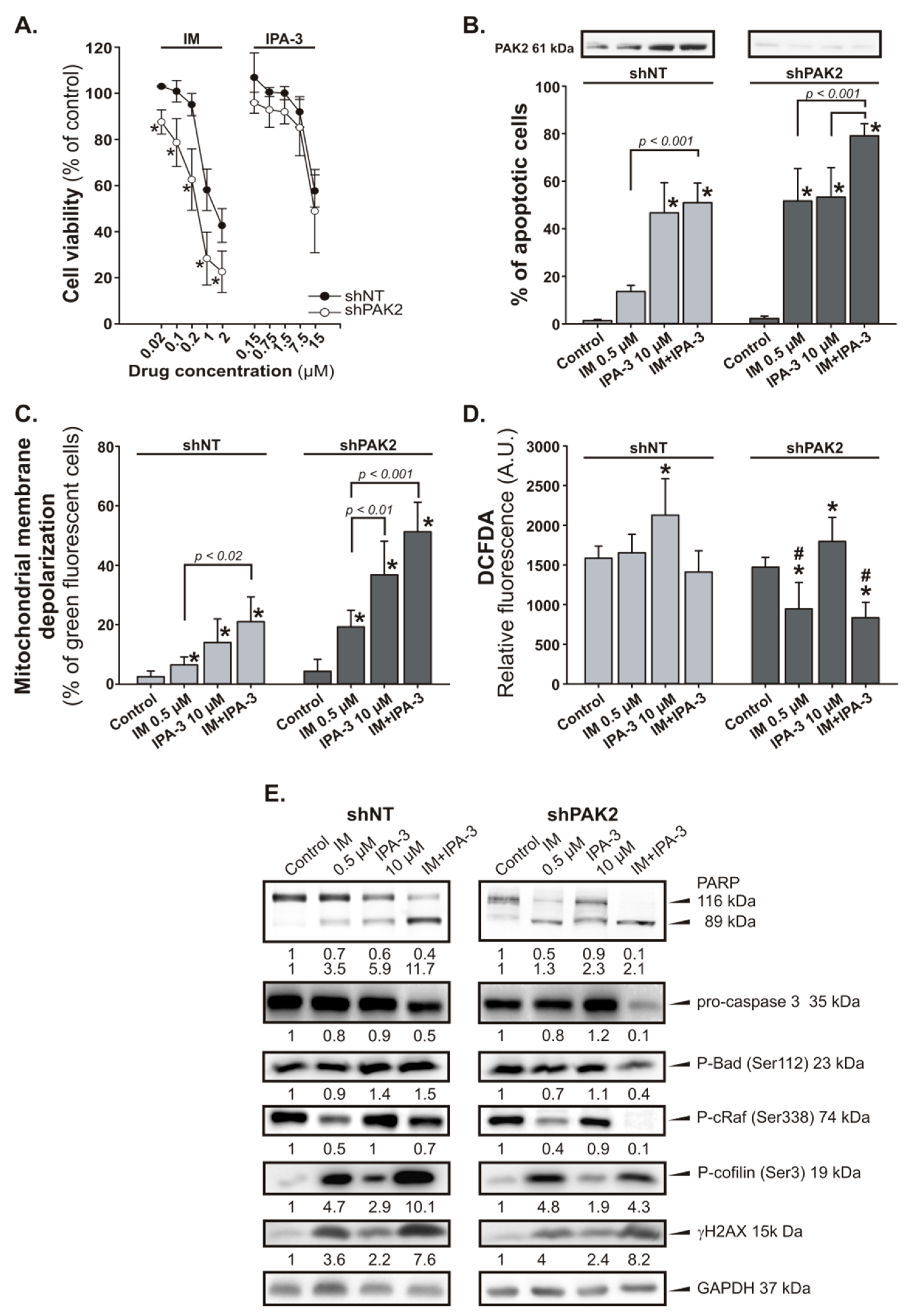
2.4. Response of PAK2 Silenced 32DclBCR-ABL1 Cells to IM
3. Discussion
4. Material and Methods
4.1. Patient Samples and Cell Culture
4.2. Drugs
4.3. 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromine (MTT) Assay
4.4. Analysis of Drug Interactions
4.5. Analysis of Cell Cycle
4.6. Analysis of Apoptosis
4.7. ROS Analysis
4.8. Oxidative DNA Damage
4.9. Immunoblotting
4.10. Measurement of Mitochondrial Membrane Potential (ΔΨm)
4.11. Colony-Forming Assay
4.12. shRNA Transfection
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Melo, J.V.; Barnes, D.J. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat. Rev. Cancer 2007, 7, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Marley, S.B.; Gordon, M.Y. Chronic myeloid leukaemia, stem cell derived but progenitor cell driven. Clin. Sci. (Lond.) 2005, 109, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Perrotti, D.; Jamieson, C.; Goldman, J.; Skorski, T. Chronic myeloid leukemia, mechanisms of blastic transformation. J. Clin. Invest. 2010, 120, 2254–2264. [Google Scholar] [CrossRef] [PubMed]
- Foroni, L.; Gerrard, G.; Nna, E.; Khorashad, J.S.; Stevens, D.; Swale, B.; Milojkovic, D.; Reid, A.; Goldman, J.; Marin, D. Technical aspects and clinical applications of measuring BCR-ABL1 transcripts number in chronic myeloid leukemia. Am. J. Hematol. 2009, 84, 517–522. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Hughes, T.P. Predicting the response of CML patients to tyrosine kinase inhibitor therapy. Curr. Hematol. Malig. Rep. 2011, 6, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Copland, M.; Hamilton, A.; Elrick, L.J.; Baird, J.W.; Allan, E.K.; Jordanides, N.; Barow, M.; Mountford, J.C.; Holyoake, T.L. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML, but does not eliminate the quiescent fraction. Blood 2006, 107, 4532–4539. [Google Scholar] [CrossRef]
- Jorgensen, H.G.; Allan, E.K.; Jordanides, N.E.; Mountford, J.C.; Holyoake, T.L. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 2007, 109, 4016–4019. [Google Scholar] [CrossRef] [Green Version]
- Chomel, J.C.; Bonnet, M.L.; Sorel, N.; Bertrand, A.; Meunier, M.C.; Fichelson, S.; Melkus, M.; Bennaceur-Griscelli, A.; Guilhot, F.; Turhan, A.G. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood 2011, 118, 3657–3660. [Google Scholar] [CrossRef]
- Chu, S.; McDonald, T.; Lin, A.; Chakraborty, S.; Huang, Q.; Snyder, D.S.; Bhatia, R. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood 2011, 118, 5565–5572. [Google Scholar] [CrossRef] [Green Version]
- Mahon, F.X.; Rea, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years, the prospective, multicentre Stop Imatinib (STIM) trial. Lancet. Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Skorski, T. Chronic myeloid leukemia cells refractory/resistant to tyrosine kinase inhibitors are genetically unstable and may cause relapse and malignant progression to the terminal disease state. Leuk. Lymphoma 2011, 52 (Suppl. 1), 23–29. [Google Scholar] [CrossRef]
- Santos, F.P.; Quintas-Cardama, A. New drugs for chronic myelogenous leukemia. Curr. Hematol. Malig. Rep. 2011, 6, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Shami, P.J.; Deininger, M. Evolving treatment strategies for patients newly diagnosed with chronic myeloid leukemia, the role of second-generation BCR-ABL inhibitors as first-line therapy. Leukemia 2012, 26, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Eide, C.A.; Adrian, L.T.; Tyner, J.W.; Mac Partlin, M.; Anderson, D.J.; Wise, S.C.; Smith, B.D.; Petillo, P.A.; Flynn, D.L.; Deininger, M.W.; et al. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Res. 2011, 71, 3189–3195. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.T.; Yeung, D.T.; Yeoman, A.L.; Altamura, H.K.; Jamison, B.A.; Field, C.; Hodgson, J.G.; Lustgarten, S.; Rivera, V.M.; Hughes, T.P.; et al. The impact of multiple low-level BCR-ABL1 mutations on response to ponatinib. Blood 2016, 127, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.C.; Cortes, J.; Galbincea, J.; Reddy, N.; Breeden, M.; Jabbour, E.; Luthra, R.; Jones, D. Rapid clonal shifts in response to kinase inhibitor therapy in chronic myelogenous leukemia are identified by quantitation mutation assays. Cancer Sci. 2010, 101, 2005–2010. [Google Scholar] [CrossRef] [Green Version]
- Skorski, T. BCR-ABL1 Kinase, Hunting an Elusive Target with New Weapons. Chem. Biol. 2011, 18, 1352–1353. [Google Scholar] [CrossRef]
- Woessner, D.W.; Lim, C.S.; Deininger, M.W. Development of an effective therapy for chronic myelogenous leukemia. Cancer J. 2011, 17, 477–486. [Google Scholar] [CrossRef]
- Skorski, T.; Kanakaraj, P.; Nieborowska-Skorska, M.; Ratajczak, M.Z.; Wen, S.C.; Zon, G.; Gewirtz, A.M.; Perussia, B.; Calabretta, B. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood 1995, 86, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Skorski, T.; Bellacosa, A.; Nieborowska-Skorska, M.; Majewski, M.; Martinez, R.; Choi, J.K.; Trotta, R.; Wlodarski, P.; Perrotti, D.; Chan, T.O.; et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. Embo. J. 1997, 16, 6151–6161. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Slupianek, A.; Skorski, T. Progressive changes in the leukemogenic signaling in BCR/ABL-transformed cells. Oncogene 2000, 19, 4117–4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klejman, A.; Rushen, L.; Morrione, A.; Slupianek, A.; Skorski, T. Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene 2002, 21, 5868–5876. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.Y.; Bolton, E.; Mohi, M.G.; Morrione, A.; Neel, B.G.; Skorski, T. Phosphatidylinositol 3-kinase p85{alpha} subunit-dependent interaction with BCR/ABL-related fusion tyrosine kinases, molecular mechanisms and biological consequences. Mol. Cell Biol. 2005, 25, 8001–8008. [Google Scholar] [CrossRef] [PubMed]
- Andreu, E.J.; Lledo, E.; Poch, E.; Ivorra, C.; Albero, M.P.; Martinez-Climent, J.A.; Montiel-Duarte, C.; Rifón, J.; Pérez-Calvo, J.; Arbona, C.; et al. BCR-ABL induces the expression of Skp2 through the PI3K pathway to promote p27Kip1 degradation and proliferation of chronic myelogenous leukemia cells. Cancer Res. 2005, 65, 3264–3272. [Google Scholar] [CrossRef]
- Kharas, M.G.; Janes, M.R.; Scarfone, V.M.; Lilly, M.B.; Knight, Z.A.; Shokat, K.M.; Fruman, D.A. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J. Clin. Invest. 2008, 118, 3038–3050. [Google Scholar] [CrossRef] [Green Version]
- Sattler, M.; Mohi, M.G.; Pride, Y.B.; Quinnan, L.R.; Malouf, N.A.; Podar, K.; Gesbert, F.; Iwasaki, H.; Li, S.; Van Etten, R.A.; et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell 2002, 1, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Jin, A.; Kurosu, T.; Tsuji, K.; Mizuchi, D.; Arai, A.; Fujita, H.; Hattori, M.; Minato, N.; Miura, O. BCR/ABL and IL-3 activate Rap1 to stimulate the B-Raf/MEK/Erk and Akt signaling pathways and to regulate proliferation, apoptosis, and adhesion. Oncogene 2006, 25, 4332–4340. [Google Scholar] [CrossRef] [Green Version]
- Atfi, A.; Abecassis, L.; Bourgeade, M.F. Bcr-Abl activates the AKT/Fox O3 signalling pathway to restrict transforming growth factor-beta-mediated cytostatic signals. EMBO Rep. 2005, 6, 985–991. [Google Scholar] [CrossRef]
- Chu, S.; Li, L.; Singh, H.; Bhatia, R. BCR-tyrosine 177 plays an essential role in Ras and Akt activation and in human hematopoietic progenitor transformation in chronic myelogenous leukemia. Cancer Res. 2007, 67, 7045–7053. [Google Scholar] [CrossRef]
- Sengupta, A.; Arnett, J.; Dunn, S.; Williams, D.A.; Cancelas, J.A. Rac2 GTPase deficiency depletes BCR-ABL+ leukemic stem cells and progenitors in vivo. Blood 2010, 116, 81–84. [Google Scholar] [CrossRef]
- Thomas, E.K.; Cancelas, J.A.; Chae, H.D.; Cox, A.D.; Keller, P.J.; Perrotti, D.; Neviani, P.; Druker, B.J.; Setchell, K.D.; Zheng, Y.; et al. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell 2007, 12, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Bolton-Gillespie, E.; Schemionek, M.; Klein, H.U.; Flis, S.; Hoser, G.; Lange, T.; Nieborowska-Skorska, M.; Maier, J.; Kerstiens, L.; Koptyra, M.; et al. Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood 2013, 121, 4175–4183. [Google Scholar] [CrossRef] [PubMed]
- Flis, K.; Irvine, D.; Copland, M.; Bhatia, R.; Skorski, T. Chronic myeloid leukemia stem cells display alterations in expression of genes involved in oxidative phosphorylation. Leuk Lymphoma 2012, 53, 2474–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieborowska-Skorska, M.; Kopinski, P.K.; Ray, R.; Hoser, G.; Ngaba, D.; Flis, S.; Cramer, K.; Reddy, M.M.; Koptyra, M.; Penserga, T.; et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012, 119, 4253–4263. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Flis, S.; Skorski, T. AKT-induced reactive oxygen species generate imatinib-resistant clones emerging from chronic myeloid leukemia progenitor cells. Leukemia 2014, 28, 2416–2418. [Google Scholar] [CrossRef] [Green Version]
- Molli, P.R.; Li, D.Q.; Murray, B.W.; Rayala, S.K.; Kumar, R. PAK signaling in oncogenesis. Oncogene 2009, 28, 2545–2555. [Google Scholar] [CrossRef] [Green Version]
- Deacon, S.W.; Beeser, A.; Fukui, J.A.; Rennefahrt, U.E.; Myers, C.; Chernoff, J.; Peterson, J.R. An isoform-selective, small-molecule inhibitor targets the autoregulatory, mechanism of p21-activated kinase. Chem. Biol. 2008, 15, 322–331. [Google Scholar] [CrossRef]
- Ye, D.Z.; Field, J. PAK signaling in cancer. Cell Logist. 2012, 2, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.R.; Tolentino, J.H.; Hazlehurst, L.A. Role of STAT3 in Transformation and Drug Resistance in CML. Front. Oncol. 2012, 2, 30. [Google Scholar] [CrossRef] [Green Version]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer. 2014, 14, 13–25. [Google Scholar] [CrossRef]
- Flis, S.; Chojnacki, T. Chronic myelogenous leukemia, a still unsolved problem: pitfalls and new therapeutic possibilities. Drug Des. Devel. Ther. 2019, 13, 825–843. [Google Scholar] [CrossRef] [PubMed]
- Knaus, U.G.; Bokoch, G.M. The p21Rac/Cdc42-activated kinases (PAKs). Int. J. Biochem. Cell Biol. 1998, 30, 857–862. [Google Scholar] [CrossRef]
- Edlinger, L.; Berger-Becvar, A.; Menzl, I.; Hoermann, G.; Greiner, G.; Grundschober, E. Expansion of BCR/ABL1(+) cells requires PAK2 but not PAK1. Br. J. Haematol. 2017, 179, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Buchdunger, E.; Druker, B.J. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005, 105, 2640–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuželová, K.; Grebeňová, D.; Holoubek, A.; Röselová, P.; Obr, A. Group I PAK inhibitor IPA-3 induces cell death and affects cell adhesivity to fibronectin in human hematopoietic cells. PLoS ONE 2014, 9, e92560. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef]
- Lappalainen, P.; Drubin, D.G. Cofilin promotes rapid actin filament turnover in vivo. Nature 1997, 388, 78–82. [Google Scholar] [CrossRef]
- Li, J.; Li, Q.; Xie, C.; Zhou, H.; Wang, Y.; Zhang, N.; Shao, H.; Chan, S.C.; Peng, X.; Lin, S.C.; et al. Beta-actin is required for mitochondria clustering and ROS generation in TNF-induced, caspase-independent cell death. J. Cell Sci. 2004, 117, 4673–4680. [Google Scholar] [CrossRef]
- Prudent, R.; Demoncheaux, N.; Diemer, H.; Collin-Faure, V.; Kapur, R.; Paublant, F.; Lafanechère, L.; Cianférani, S.; Rabilloud, T. A quantitative proteomic analysis of cofilin phosphorylation in myeloid cells and its modulation using the LIM kinase inhibitor Pyr1. PLoS ONE 2018, 3, e0208979. [Google Scholar] [CrossRef]
- Ritchey, L.; Chakrabarti, R. Aurora A kinase modulates actin cytoskeleton through phosphorylation of Cofilin, Implication in the mitotic process. Biochim. Biophys. Acta. 2014, 1843, 2719–2729. [Google Scholar] [CrossRef]
- Chang, C.Y.; Leu, J.D.; Lee, Y.J. The actin depolymerizing factor (ADF)/cofilin signaling pathway and DNA damage responses in cancer. Int. J. Mol. Sci. 2015, 16, 4095–4120. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieborowska-Skorska, M.; Wasik, M.A.; Slupianek, A.; Salomoni, P.; Kitamura, T.; Calabretta, B.; Skorski, T. Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J. Exp. Med. 1999, 189, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Invest. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flis, S.; Bratek, E.; Chojnacki, T.; Piskorek, M.; Skorski, T. Simultaneous Inhibition of BCR-ABL1 Tyrosine Kinase and PAK1/2 Serine/Threonine Kinase Exerts Synergistic Effect against Chronic Myeloid Leukemia Cells. Cancers 2019, 11, 1544. https://doi.org/10.3390/cancers11101544
Flis S, Bratek E, Chojnacki T, Piskorek M, Skorski T. Simultaneous Inhibition of BCR-ABL1 Tyrosine Kinase and PAK1/2 Serine/Threonine Kinase Exerts Synergistic Effect against Chronic Myeloid Leukemia Cells. Cancers. 2019; 11(10):1544. https://doi.org/10.3390/cancers11101544
Chicago/Turabian StyleFlis, Sylwia, Ewelina Bratek, Tomasz Chojnacki, Marlena Piskorek, and Tomasz Skorski. 2019. "Simultaneous Inhibition of BCR-ABL1 Tyrosine Kinase and PAK1/2 Serine/Threonine Kinase Exerts Synergistic Effect against Chronic Myeloid Leukemia Cells" Cancers 11, no. 10: 1544. https://doi.org/10.3390/cancers11101544
APA StyleFlis, S., Bratek, E., Chojnacki, T., Piskorek, M., & Skorski, T. (2019). Simultaneous Inhibition of BCR-ABL1 Tyrosine Kinase and PAK1/2 Serine/Threonine Kinase Exerts Synergistic Effect against Chronic Myeloid Leukemia Cells. Cancers, 11(10), 1544. https://doi.org/10.3390/cancers11101544