The Complexities of Metastasis

 , and
, and 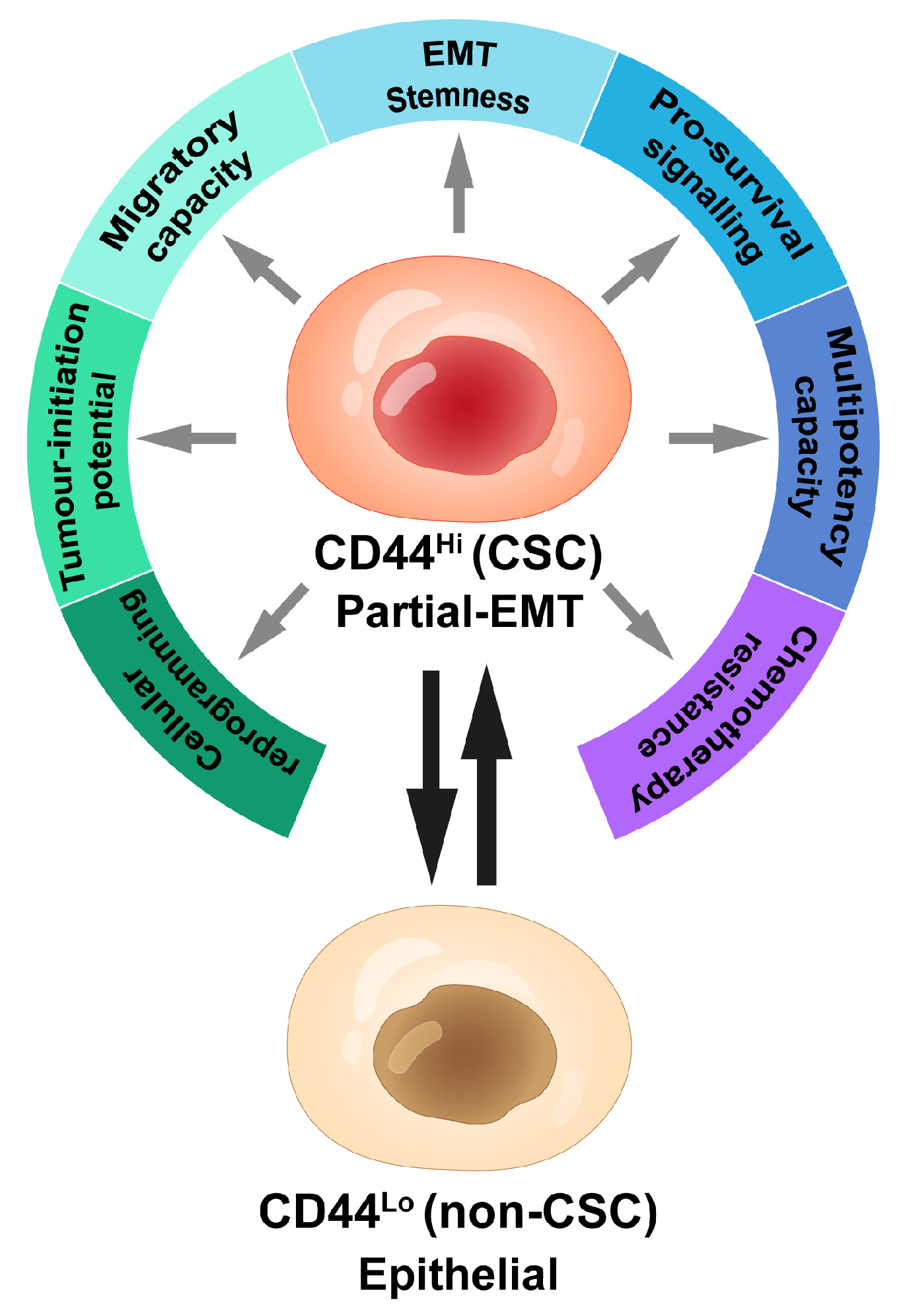{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Cancer Cell Heterogeneity: A Hierarchical Matter?
1.1. Cancer Origin and Evolution
1.2. A Cancer Cell Hierarchy
1.3. CD44: Defining Aggressive Cancer Cells
1.3.1. CD44Lo versus CD44Hi Cells: Epithelial versus Mesenchymal Cell States
1.3.2. Novel Markers to Define Metastatic Cells
2. Cancer Cell Plasticity: Shaping Metastatic Fitness
3. The Seed, the Journey, and the Soil: The Metastatic Cascade
3.1. Entering the Circulation, Off They Go
3.2. In Transit: Better Together
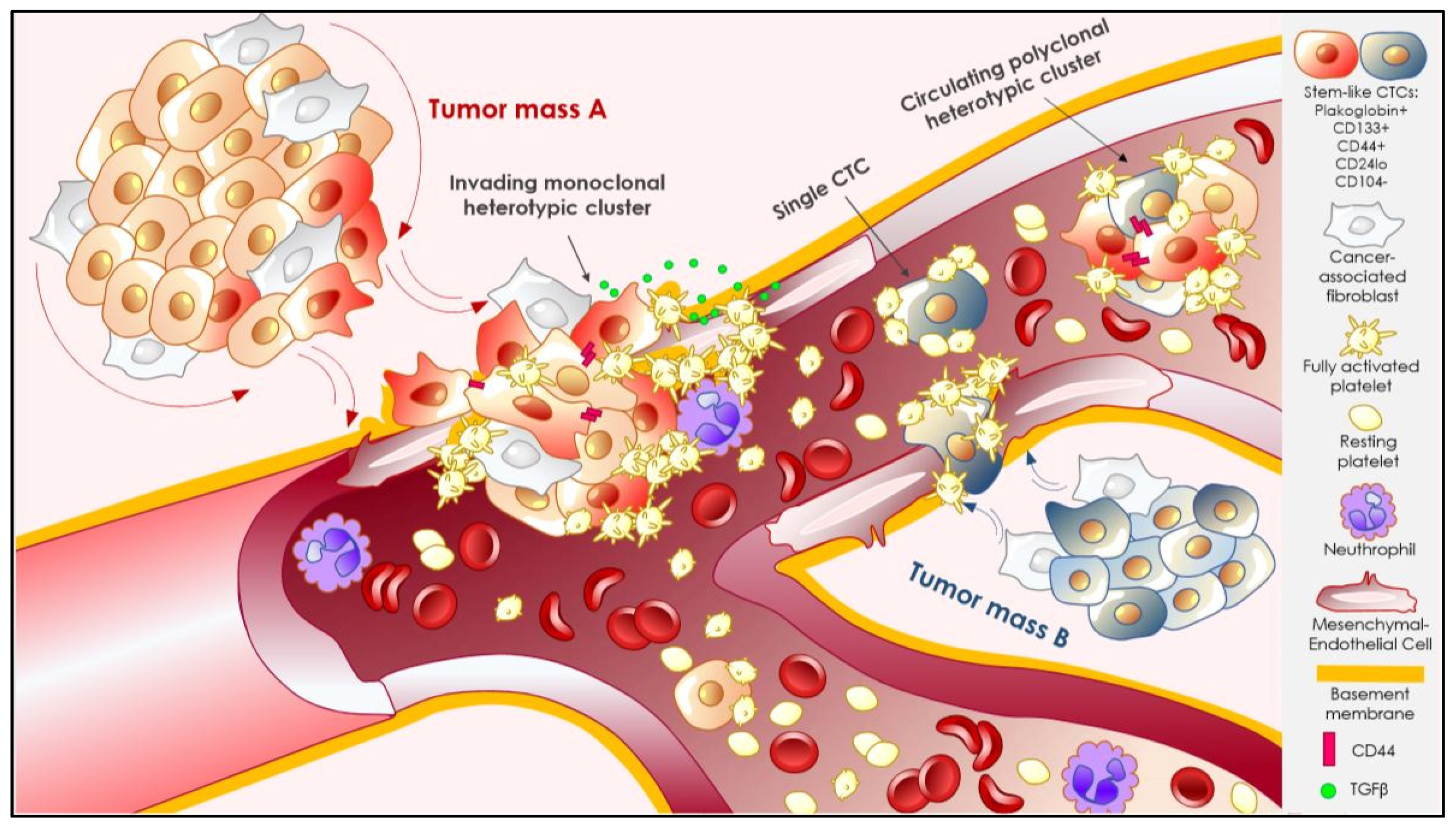
3.2.1. CTC Clustering
3.2.2. Interactions That Matter: Heterotypic Clustering
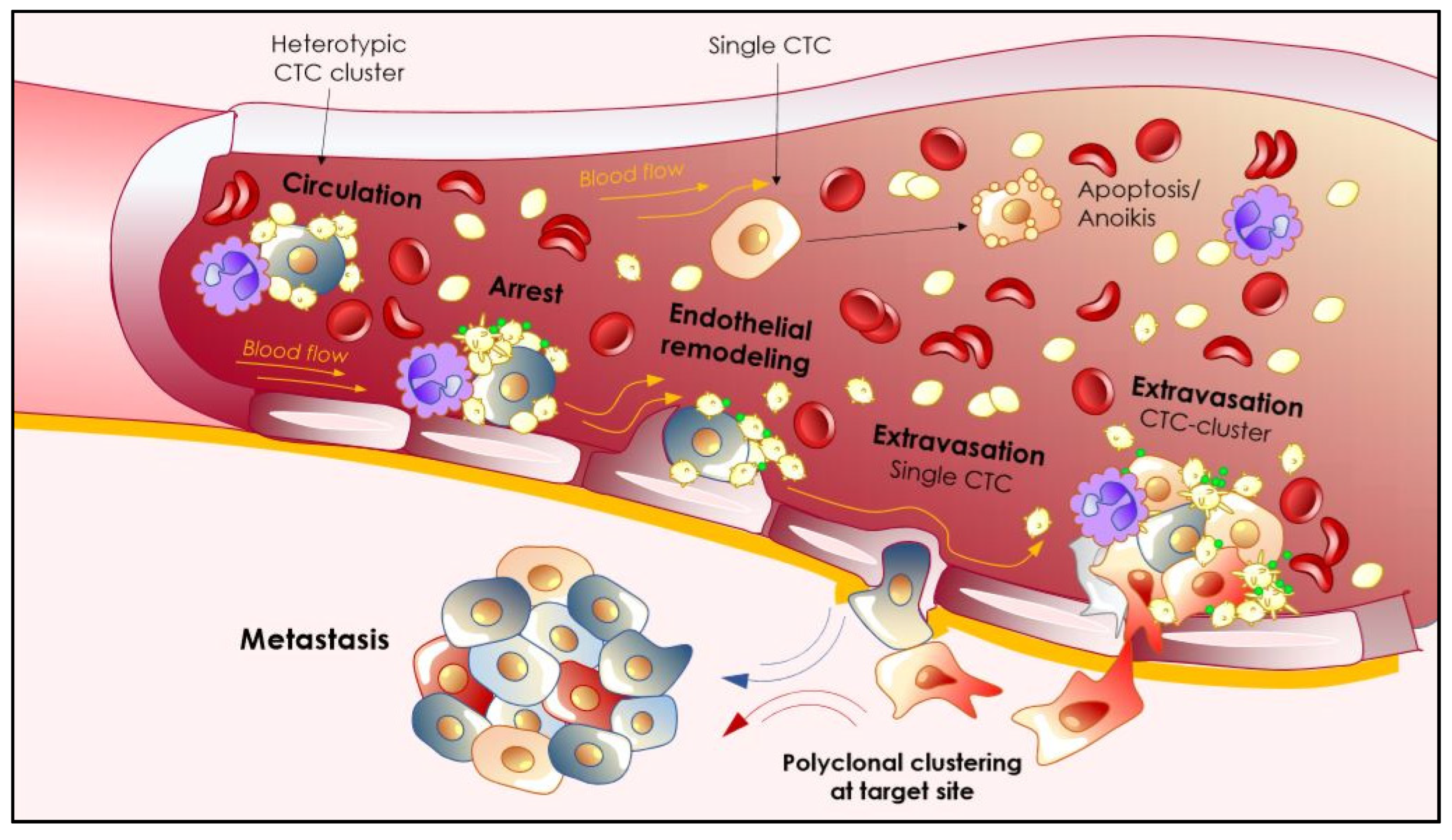
3.2.3. Going with the Flow: Biomechanics of CTCs Extravasation
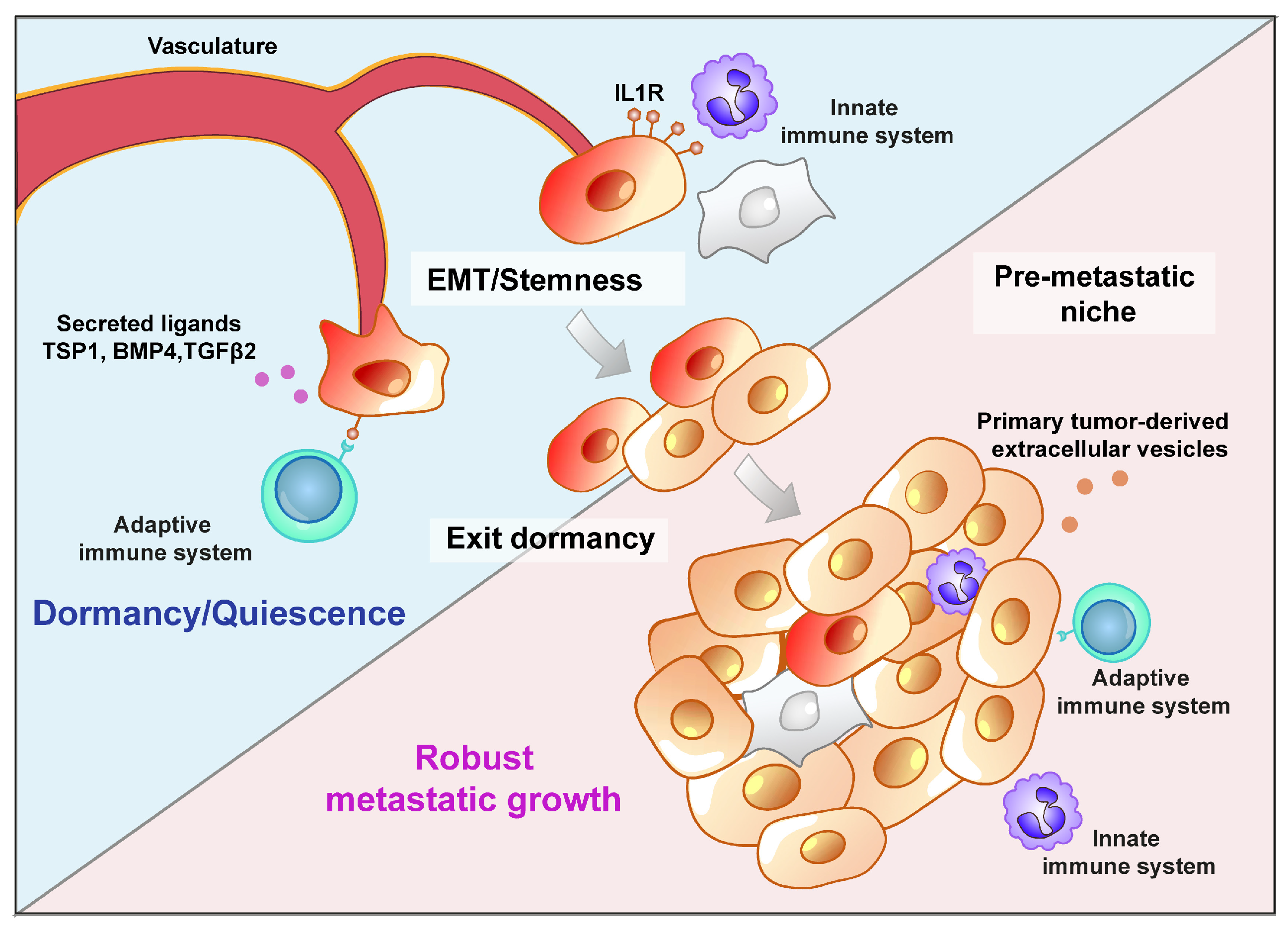
4. Secondary Organ Colonization: Shedders or Seeders?
Metastatic Niche: A Driving Force or a Barrier?
5. Conclusions
Funding
Conflicts of Interest
References
- Weinstein, B.T.; Lavrentovich, M.O.; Möbius, W.; Murray, A.W.; Nelson, D.R. Genetic drift and selection in many-allele range expansions. PLoS Comput. Biol. 2017, 13, e1005866. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Rubin, H.; Rubin, A.L. Phenotypic selection as the biological mode of epigenetic conversion and reversion in cell transformation. Proc. Natl. Acad. Sci. USA 2018, 115, E725–E732. [Google Scholar] [CrossRef] [PubMed]
- Rybinski, B.; Yun, K. Addressing intra-tumoral heterogeneity and therapy resistance. Oncotarget 2016, 7, 72322–72342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.-J.; Ingram, P.N.; Yang, K.; Coffman, L.; Iyengar, M.; Bai, S.; Thomas, D.G.; Yoon, E.; Buckanovich, R.J. Identifying an ovarian cancer cell hierarchy regulated by bone morphogenetic protein 2. Proc. Natl. Acad. Sci. USA 2015, 112, E6882–E6888. [Google Scholar] [CrossRef] [Green Version]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.S.; Huang, J.; Guo, C.; Garraway, I.P.; Witte, O.N. Identification of a cell of origin for human prostate cancer. Science 2010, 329, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Vanharanta, S.; Massagué, J. Origins of Metastatic Traits. Cancer Cell 2013, 24, 410–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Neaves, W.B. Normal Stem Cells and Cancer Stem Cells: The Niche Matters: Figure 1. Cancer Res. 2006, 66, 4553–4557. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [Green Version]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef]
- Merino, D.; Weber, T.S.; Serrano, A.; Vaillant, F.; Liu, K.; Pal, B.; Di Stefano, L.; Schreuder, J.; Lin, D.; Chen, Y.; et al. Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat. Commun. 2019, 10, 766. [Google Scholar] [CrossRef]
- Wagenblast, E.; Soto, M.; Gutiérrez-Ángel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.V.; Cox, C.L.; Eirew, P.; Knapp, D.J.H.F.; Pellacani, D.; Kannan, N.; Carles, A.; Moksa, M.; Balani, S.; Shah, S.; et al. DNA barcoding reveals diverse growth kinetics of human breast tumour subclones in serially passaged xenografts. Nat. Commun. 2014, 5, 5871. [Google Scholar] [CrossRef] [Green Version]
- Maleki, M.; Ghanbarvand, F.; Behvarz, M.R.; Ejtemaei, M.; Ghadirkhomi, E. Comparison of Mesenchymal Stem Cell Markers in Multiple Human Adult Stem Cells. Int. J. Stem Cells 2014, 7, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryczek, I.; Liu, S.; Roh, M.; Vatan, L.; Szeliga, W.; Wei, S.; Banerjee, M.; Mao, Y.; Kotarski, J.; Wicha, M.S.; et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer 2012, 130, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friel, A.M.; Roberts, D.J.; Seiden, M.V.; Scadden, D.T.; Rueda, B.R.; et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Boiko, A.D.; Razorenova, O.V.; van de Rijn, M.; Swetter, S.M.; Johnson, D.L.; Ly, D.P.; Butler, P.D.; Yang, G.P.; Joshua, B.; Kaplan, M.J.; et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 2010, 466, 133–137. [Google Scholar] [CrossRef]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.W.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.K.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Lin, J.; Ding, D. The prognostic role of the cancer stem cell marker CD44 in ovarian cancer: A meta-analysis. Cancer Cell Int. 2017, 17, 8. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Dobbin, Z.C.; Katre, A.A.; Steg, A.D.; Erickson, B.K.; Shah, M.M.; Alvarez, R.D.; Conner, M.G.; Schneider, D.; Chen, D.; Landen, C.N. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 2014, 5, 8750–8764. [Google Scholar] [CrossRef]
- Garcia-Peydro, M.; Fuentes, P.; Mosquera, M.; Garcia-Leon, M.J.; Alcain, J.; Rodriguez, A.; Garcia de Miguel, P.; Menendez, P.; Weijer, K.; Spits, H.; et al. The NOTCH1/CD44 axis drives pathogenesis in a T cell acute lymphoblastic leukemia model. J. Clin. Investig. 2018, 128, 2802–2818. [Google Scholar] [CrossRef] [PubMed]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martínez-Sáez, E.; Prudkin, L.; et al. TGF-β Receptor Inhibitors Target the CD44high/Id1high Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Z.; Wong, S.K.-M.; Tin, V.P.-C.; Ho, K.-Y.; Wang, J.; Sham, M.-H.; Wong, M.P. Lung cancer tumorigenicity and drug resistance are maintained through ALDH(hi)CD44(hi) tumor initiating cells. Oncotarget 2013, 4, 1698–1711. [Google Scholar] [CrossRef]
- Bansal, N.; Davis, S.; Tereshchenko, I.; Budak-Alpdogan, T.; Zhong, H.; Stein, M.N.; Kim, I.Y.; DiPaola, R.S.; Bertino, J.R.; Sabaawy, H.E. Enrichment of human prostate cancer cells with tumor initiating properties in mouse and zebrafish xenografts by differential adhesion. Prostate 2014, 74, 187–200. [Google Scholar] [CrossRef]
- Shen, S.; Yang, W.; Wang, Z.; Lei, X.; Xu, L.; Wang, Y.; Wang, L.; Huang, L.; Yu, Z.; Zhang, X.; et al. Tumor-Initiating Cells Are Enriched in CD44hi Population in Murine Salivary Gland Tumor. PLoS ONE 2011, 6, e23282. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Bourguignon, L.Y.W.; Gunja-Smith, Z.; Iida, N.; Zhu, H.B.; Young, L.J.T.; Muller, W.J.; Cardiff, R.D. CD44v3,8-10 is involved in cytoskeleton-mediated tumor cell migration and matrix metalloproteinase (MMP-9) association in metastatic breast cancer cells. J. Cell Physiol. 1998, 176, 206–215. [Google Scholar] [CrossRef]
- Zeilstra, J.; Joosten, S.P.J.; van Andel, H.; Tolg, C.; Berns, A.; Snoek, M.; van de Wetering, M.; Spaargaren, M.; Clevers, H.; Pals, S.T. Stem cell CD44v isoforms promote intestinal cancer formation in Apc(min) mice downstream of Wnt signaling. Oncogene 2014, 33, 665–670. [Google Scholar] [CrossRef]
- Li, Z.; Chen, K.; Jiang, P.; Zhang, X.; Li, X.; Li, Z. CD44v/CD44s expression patterns are associated with the survival of pancreatic carcinoma patients. Diagn. Pathol. 2014, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-Y.; Guo, M.; Yang, L.-Q.; Zhou, F.; Yu, C.; Wang, A.; Pang, T.-H.; Wu, H.-Y.; Zou, X.-P.; Zhang, W.-J.; et al. Regulation of CD44v6 expression in gastric carcinoma by the IL-6/STAT3 signaling pathway and its clinical significance. Oncotarget 2017, 8, 45848–45861. [Google Scholar] [CrossRef] [PubMed]
- Osmani, N.; Follain, G.; Garcia Leon, M.J.; Lefebvre, O.; Busnelli, I.; Larnicol, A.; Harlepp, S.; Goetz, J.G. Metastatic Tumor Cells Exploit Their Adhesion Repertoire to Counteract Shear Forces during Intravascular Arrest. Cell Rep. 2019, 28, 2491–2500 e2495. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef]
- Larsen, J.E.; Nathan, V.; Osborne, J.K.; Farrow, R.K.; Deb, D.; Sullivan, J.P.; Dospoy, P.D.; Augustyn, A.; Hight, S.K.; Sato, M.; et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Investig. 2016, 126, 3219–3235. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Preca, B.T.; Bajdak, K.; Mock, K.; Sundararajan, V.; Pfannstiel, J.; Maurer, J.; Wellner, U.; Hopt, U.T.; Brummer, T.; Brabletz, S.; et al. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int. J. Cancer. 2015, 137, 2566–2577. [Google Scholar] [CrossRef]
- Jolly, M.K.; Preca, B.T.; Tripathi, S.C.; Jia, D.; George, J.T.; Hanash, S.M.; Brabletz, T.; Stemmler, M.P.; Maurer, J.; Levine, H. Interconnected feedback loops among ESRP1, HAS2, and CD44 regulate epithelial-mesenchymal plasticity in cancer. APL Bioeng. 2018, 2, 031908. [Google Scholar] [CrossRef]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef]
- Shen, M.; Xu, Z.; Xu, W.; Jiang, K.; Zhang, F.; Ding, Q.; Xu, Z.; Chen, Y. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 149. [Google Scholar]
- Ponnusamy, L.; Mahalingaiah, P.K.S.; Chang, Y.-W.; Singh, K.P. Role of cellular reprogramming and epigenetic dysregulation in acquired chemoresistance in breast cancer. Cancer Drug Resist. 2019. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liao, R.; Li, D.; Sun, J. Induced cancer stem cells generated by radiochemotherapy and their therapeutic implications. Oncotarget 2017, 8, 17301–17312. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Wilde, A.; Fouquier d’Herouel, A.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.A.; Huang, S. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef]
- Soundararajan, R.; Paranjape, A.N.; Barsan, V.; Chang, J.T.; Mani, S.A. A novel embryonic plasticity gene signature that predicts metastatic competence and clinical outcome. Sci. Rep. 2015, 5, 11766. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hong, S.J.; Park, J.Y.; Park, J.H.; Yu, Y.S.; Park, S.Y.; Lim, E.K.; Choi, K.Y.; Lee, E.K.; Paik, S.S.; et al. Epithelial-mesenchymal transition gene signature to predict clinical outcome of hepatocellular carcinoma. Cancer Sci. 2010, 101, 1521–1528. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 2015, 6, 6139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Huang, B.; Lu, M.; Mani, S.A.; Levine, H.; Ben-Jacob, E. Towards elucidating the connection between epithelial-mesenchymal transitions and stemness. J. R. Soc. Interface 2014, 11, 20140962. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2019, 194. [Google Scholar] [CrossRef]
- Yu, M.; Stott, S.; Toner, M.; Maheswaran, S.; Haber, D.A. Circulating tumor cells: Approaches to isolation and characterization. J. Cell Biol. 2011, 192, 373–382. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Pantel, K. Circulating tumor cells: Liquid biopsy of cancer. Clin. Chem. 2013, 59, 110–118. [Google Scholar]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Weiss, L. Metastatic inefficiency. Adv. Cancer Res. 1990, 54, 159–211. [Google Scholar]
- Blazejczyk, A.; Papiernik, D.; Porshneva, K.; Sadowska, J.; Wietrzyk, J. Endothelium and cancer metastasis: Perspectives for antimetastatic therapy. Pharmacol. Rep. 2015, 67, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngoc, K.V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C.; Yaswen, P.; Werb, Z.; Ewald, A.J. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, E2595–E2604. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Yang, L.; Lin, P.C. Mechanisms that drive inflammatory tumor microenvironment, tumor heterogeneity, and metastatic progression. Semin. Cancer Biol. 2017, 47, 185–195. [Google Scholar] [CrossRef]
- Zervantonakis, I.K.; Hughes-Alford, S.K.; Charest, J.L.; Condeelis, J.S.; Gertler, F.B.; Kamm, R.D. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. USA 2012, 109, 13515–13520. [Google Scholar] [CrossRef] [Green Version]
- Spicer, J.D.; McDonald, B.; Cools-Lartigue, J.J.; Chow, S.C.; Giannias, B.; Kubes, P.; Ferri, L.E. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer Res. 2012, 72, 3919–3927. [Google Scholar] [CrossRef]
- Gros, A.; Syvannarath, V.; Lamrani, L.; Ollivier, V.; Loyau, S.; Goerge, T.; Nieswandt, B.; Jandrot-Perrus, M.; Ho-Tin-Noe, B. Single platelets seal neutrophil-induced vascular breaches via GPVI during immune-complex-mediated inflammation in mice. Blood 2015, 126, 1017–1026. [Google Scholar] [CrossRef]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Prandoni, P. Venous thromboembolism risk and management in women with cancer and thrombophilia. Gend. Med. 2005, 2 (Suppl. A), S28–S34. [Google Scholar] [CrossRef]
- Varki, A. Trousseau’s syndrome: Multiple definitions and multiple mechanisms. Blood 2007, 110, 1723–1729. [Google Scholar] [CrossRef]
- Miyashita, T.; Tajima, H.; Makino, I.; Nakagawara, H.; Kitagawa, H.; Fushida, S.; Harmon, J.W.; Ohta, T. Metastasis-promoting role of extravasated platelet activation in tumor. J. Surg. Res. 2015, 193, 289–294. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, E3053–E3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Halabi, N.; Guerrouahen, B.S.; Rafii, S.; Rafii, A. Breast cancer cells promote a notch-dependent mesenchymal phenotype in endothelial cells participating to a pro-tumoral niche. J. Transl. Med. 2015, 13, 27. [Google Scholar] [CrossRef]
- Kontomanolis, E.; Panteliadou, M.; Giatromanolaki, A.; Pouliliou, S.; Efremidou, E.; Limberis, V.; Galazios, G.; Sivridis, E.; Koukourakis, M.I. Delta-like ligand 4 (DLL4) in the plasma and neoplastic tissues from breast cancer patients: Correlation with metastasis. Med. Oncol. 2014, 31, 945. [Google Scholar] [CrossRef]
- Mailhos, C.; Modlich, U.; Lewis, J.; Harris, A.; Bicknell, R.; Ish-Horowicz, D. Delta4, an endothelial specific notch ligand expressed at sites of physiological and tumor angiogenesis. Differentiation 2001, 69, 135–144. [Google Scholar] [CrossRef]
- Patel, N.S.; Li, J.L.; Generali, D.; Poulsom, R.; Cranston, D.W.; Harris, A.L. Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res. 2005, 65, 8690–8697. [Google Scholar] [CrossRef] [PubMed]
- Reedijk, M.; Odorcic, S.; Chang, L.; Zhang, H.; Miller, N.; McCready, D.R.; Lockwood, G.; Egan, S.E. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005, 65, 8530–8537. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Ding, B.S.; Guo, P.; Lee, S.B.; Butler, J.M.; Casey, S.C.; Simons, M.; Tam, W.; Felsher, D.W.; Shido, K.; et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 2014, 25, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef] [PubMed]
- Sonoshita, M.; Aoki, M.; Fuwa, H.; Aoki, K.; Hosogi, H.; Sakai, Y.; Hashida, H.; Takabayashi, A.; Sasaki, M.; Robine, S.; et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell 2011, 19, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Rafii, S.; Rafii, A. Endothelial cells provide a notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS ONE 2014, 9, e112424. [Google Scholar] [CrossRef]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef]
- Fidler, I.J. The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur. J. Cancer 1973, 9, 223–227. [Google Scholar] [CrossRef]
- Liotta, L.A.; Saidel, M.G.; Kleinerman, J. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res. 1976, 36, 889–894. [Google Scholar]
- Molnar, B.; Ladanyi, A.; Tanko, L.; Sreter, L.; Tulassay, Z. Circulating tumor cell clusters in the peripheral blood of colorectal cancer patients. Clin. Cancer Res. 2001, 7, 4080–4085. [Google Scholar] [PubMed]
- Cho, E.H.; Wendel, M.; Luttgen, M.; Yoshioka, C.; Marrinucci, D.; Lazar, D.; Schram, E.; Nieva, J.; Bazhenova, L.; Morgan, A.; et al. Characterization of circulating tumor cell aggregates identified in patients with epithelial tumors. Phys. Biol. 2012, 9, 016001. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Taftaf, R.; Kawaguchi, M.; Chang, Y.F.; Chen, W.; Entenberg, D.; Zhang, Y.; Gerratana, L.; Huang, S.; Patel, D.B.; et al. Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov. 2019, 9, 96–113. [Google Scholar] [CrossRef]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [Green Version]
- Entenberg, D.; Voiculescu, S.; Guo, P.; Borriello, L.; Wang, Y.; Karagiannis, G.S.; Jones, J.; Baccay, F.; Oktay, M.; Condeelis, J. A permanent window for the murine lung enables high-resolution imaging of cancer metastasis. Nat. Methods 2018, 15, 73–80. [Google Scholar] [CrossRef]
- Li, D.; Zhou, J.; Wang, L.; Shin, M.E.; Su, P.; Lei, X.; Kuang, H.; Guo, W.; Yang, H.; Cheng, L.; et al. Integrated biochemical and mechanical signals regulate multifaceted human embryonic stem cell functions. J. Cell Biol. 2010, 191, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Bennett, S.A.; Wang, L. Role of E-cadherin and other cell adhesion molecules in survival and differentiation of human pluripotent stem cells. Cell Adh. Migr. 2012, 6, 59–70. [Google Scholar] [CrossRef]
- Pieters, T.; van Roy, F. Role of cell-cell adhesion complexes in embryonic stem cell biology. J. Cell Sci. 2014, 127, 2603–2613. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112 e114. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.H.; Zhou, H.C.; Zhang, C.; Shang, L.R.; Zhang, L.; Xu, J.; Zheng, L.; Yuan, Y.; Guo, R.P.; Jia, W.H.; et al. A novel vascular pattern promotes metastasis of hepatocellular carcinoma in an epithelial-mesenchymal transition-independent manner. Hepatology 2015, 62, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer cell invasion and EMT marker expression: A three-dimensional study of the human cancer-host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mu, Z.; Chervoneva, I.; Austin, L.; Ye, Z.; Rossi, G.; Palazzo, J.P.; Sun, C.; Abu-Khalaf, M.; Myers, R.E.; et al. Longitudinally collected CTCs and CTC-clusters and clinical outcomes of metastatic breast cancer. Breast Cancer Res. Treat. 2017, 161, 83–94. [Google Scholar] [CrossRef]
- Bocci, F.; Kumar Jolly, M.; Onuchic, J.N. A biophysical model uncovers the size distribution of migrating cell clusters across cancer types. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Wei, D.; Zeng, X.; Yang, Z.; Zhou, Q.; Weng, X.; He, H.; Gu, Z.; Wei, X. Visualizing Interactions of Circulating Tumor Cell and Dendritic Cell in the Blood Circulation Using In Vivo Imaging Flow Cytometry. IEEE Trans. Biomed. Eng. 2019. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, Z.; Xu, S.; Li, X.; Yang, X.; Jin, P.; Liu, Y.; Zhou, X.; Zhang, T.; Gong, C.; et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J. Exp. Med. 2019, 216, 688–703. [Google Scholar] [CrossRef]
- Duda, D.G.; Duyverman, A.M.; Kohno, M.; Snuderl, M.; Steller, E.J.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [Green Version]
- Baeriswyl, V.; Christofori, G. The angiogenic switch in carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337. [Google Scholar] [CrossRef]
- Erpenbeck, L.; Schon, M.P. Deadly allies: The fatal interplay between platelets and metastasizing cancer cells. Blood 2010, 115, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Gasic, G.J.; Gasic, T.B.; Stewart, C.C. Antimetastatic effects associated with platelet reduction. Proc. Natl. Acad. Sci. USA 1968, 61, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Hynes, R.O. The initial hours of metastasis: The importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Mangin, P.; Lanza, F.; Gachet, C. Platelets in cancer. From basic research to therapeutic implications. Hamostaseologie 2015, 35, 325–336. [Google Scholar] [CrossRef]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef] [Green Version]
- Chew, H.K.; Wun, T.; Harvey, D.J.; Zhou, H.; White, R.H. Incidence of venous thromboembolism and the impact on survival in breast cancer patients. J. Clin. Oncol. 2007, 25, 70–76. [Google Scholar] [CrossRef]
- Jain, S.; Zuka, M.; Liu, J.; Russell, S.; Dent, J.; Guerrero, J.A.; Forsyth, J.; Maruszak, B.; Gartner, T.K.; Felding-Habermann, B.; et al. Platelet glycoprotein Ib alpha supports experimental lung metastasis. Proc. Natl. Acad. Sci. USA 2007, 104, 9024–9028. [Google Scholar] [CrossRef]
- Jain, S.; Russell, S.; Ware, J. Platelet glycoprotein VI facilitates experimental lung metastasis in syngenic mouse models. J. Thromb Haemost. 2009, 7, 1713–1717. [Google Scholar] [CrossRef]
- Jurasz, P.; Alonso-Escolano, D.; Radomski, M.W. Platelet--cancer interactions: Mechanisms and pharmacology of tumour cell-induced platelet aggregation. Br. J. Pharmacol. 2004, 143, 819–826. [Google Scholar] [CrossRef]
- Karpatkin, S.; Ambrogio, C.; Pearlstein, E. The role of tumor-induced platelet aggregation, platelet adhesion and adhesive proteins in tumor metastasis. Prog. Clin. Biol. Res. 1988, 283, 585–606. [Google Scholar]
- Karpatkin, S.; Pearlstein, E.; Ambrogio, C.; Coller, B.S. Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J. Clin. Investig. 1988, 81, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Mannel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar] [PubMed]
- Katagiri, Y.; Hayashi, Y.; Baba, I.; Suzuki, H.; Tanoue, K.; Yamazaki, H. Characterization of platelet aggregation induced by the human melanoma cell line HMV-I: Roles of heparin, plasma adhesive proteins, and tumor cell membrane proteins. Cancer Res. 1991, 51, 1286–1293. [Google Scholar] [PubMed]
- Alonso-Escolano, D.; Strongin, A.Y.; Chung, A.W.; Deryugina, E.I.; Radomski, M.W. Membrane type-1 matrix metalloproteinase stimulates tumour cell-induced platelet aggregation: Role of receptor glycoproteins. Br. J. Pharmacol. 2004, 141, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Brakenhoff, R.H. Dissecting the metastatic cascade. Nat. Rev. Cancer 2004, 4, 448–456. [Google Scholar] [CrossRef]
- Echtler, K.; Konrad, I.; Lorenz, M.; Schneider, S.; Hofmaier, S.; Plenagl, F.; Stark, K.; Czermak, T.; Tirniceriu, A.; Eichhorn, M.; et al. Platelet GPIIb supports initial pulmonary retention but inhibits subsequent proliferation of melanoma cells during hematogenic metastasis. PLoS ONE 2017, 12, e0172788. [Google Scholar] [CrossRef]
- Erpenbeck, L.; Nieswandt, B.; Schon, M.; Pozgajova, M.; Schon, M.P. Inhibition of platelet GPIb alpha and promotion of melanoma metastasis. J. Investig. Dermatol. 2010, 130, 576–586. [Google Scholar] [CrossRef]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Papa, A.L.; Jiang, A.; Korin, N.; Chen, M.B.; Langan, E.T.; Waterhouse, A.; Nash, E.; Caroff, J.; Graveline, A.; Vernet, A.; et al. Platelet decoys inhibit thrombosis and prevent metastatic tumor formation in preclinical models. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Amirkhosravi, A.; Mousa, S.A.; Amaya, M.; Blaydes, S.; Desai, H.; Meyer, T.; Francis, J.L. Inhibition of tumor cell-induced platelet aggregation and lung metastasis by the oral GpIIb/IIIa antagonist XV454. Thromb Haemost. 2003, 90, 549–554. [Google Scholar]
- Millard, M.; Odde, S.; Neamati, N. Integrin targeted therapeutics. Theranostics 2011, 1, 154–188. [Google Scholar] [CrossRef] [PubMed]
- Safaee, M.; Clark, A.J.; Ivan, M.E.; Oh, M.C.; Bloch, O.; Sun, M.Z.; Oh, T.; Parsa, A.T. CD97 is a multifunctional leukocyte receptor with distinct roles in human cancers (Review). Int. J. Oncol. 2013, 43, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Boucharaba, A.; Serre, C.M.; Gres, S.; Saulnier-Blache, J.S.; Bordet, J.C.; Guglielmi, J.; Clezardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Investig. 2004, 114, 1714–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblanc, R.; Lee, S.C.; David, M.; Bordet, J.C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clezardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood 2014, 124, 3141–3150. [Google Scholar] [CrossRef] [PubMed]
- Ward, Y.; Lake, R.; Yin, J.J.; Heger, C.D.; Raffeld, M.; Goldsmith, P.K.; Merino, M.; Kelly, K. LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res. 2011, 71, 7301–7311. [Google Scholar] [CrossRef]
- Ward, Y.; Lake, R.; Faraji, F.; Sperger, J.; Martin, P.; Gilliard, C.; Ku, K.P.; Rodems, T.; Niles, D.; Tillman, H.; et al. Platelets Promote Metastasis via Binding Tumor CD97 Leading to Bidirectional Signaling that Coordinates Transendothelial Migration. Cell Rep. 2018, 23, 808–822. [Google Scholar] [CrossRef]
- Kato, Y.; Kaneko, M.K.; Kunita, A.; Ito, H.; Kameyama, A.; Ogasawara, S.; Matsuura, N.; Hasegawa, Y.; Suzuki-Inoue, K.; Inoue, O.; et al. Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci. 2008, 99, 54–61. [Google Scholar] [CrossRef]
- Yu, L.X.; Yan, L.; Yang, W.; Wu, F.Q.; Ling, Y.; Chen, S.Z.; Tang, L.; Tan, Y.X.; Cao, D.; Wu, M.C.; et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat. Commun. 2014, 5, 5256. [Google Scholar] [CrossRef]
- Aigner, S.; Sthoeger, Z.M.; Fogel, M.; Weber, E.; Zarn, J.; Ruppert, M.; Zeller, Y.; Vestweber, D.; Stahel, R.; Sammar, M.; et al. CD24, a mucin-type glycoprotein, is a ligand for P-selectin on human tumor cells. Blood 1997, 89, 3385–3395. [Google Scholar] [CrossRef]
- Kristiansen, G.; Sammar, M.; Altevogt, P. Tumour biological aspects of CD24, a mucin-like adhesion molecule. J. Mol. Histol. 2004, 35, 255–262. [Google Scholar] [CrossRef]
- Yao, X.; Labelle, M.; Lamb, C.R.; Dugan, J.M.; Williamson, C.A.; Spencer, D.R.; Christ, K.R.; Keating, R.O.; Lee, W.D.; Paradis, G.A.; et al. Determination of 35 cell surface antigen levels in malignant pleural effusions identifies CD24 as a marker of disseminated tumor cells. Int. J. Cancer 2013, 133, 2925–2933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haemmerle, M.; Taylor, M.L.; Gutschner, T.; Pradeep, S.; Cho, M.S.; Sheng, J.; Lyons, Y.M.; Nagaraja, A.S.; Dood, R.L.; Wen, Y.; et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun. 2017, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Pilch, J.; Habermann, R.; Felding-Habermann, B. Unique ability of integrin alpha(v)beta 3 to support tumor cell arrest under dynamic flow conditions. J. Biol. Chem. 2002, 277, 21930–21938. [Google Scholar] [CrossRef]
- Frenette, P.S.; Johnson, R.C.; Hynes, R.O.; Wagner, D.D. Platelets roll on stimulated endothelium in vivo: An interaction mediated by endothelial P-selectin. Proc. Natl. Acad. Sci. USA 1995, 92, 7450–7454. [Google Scholar] [CrossRef]
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef]
- Fung, Y.C. Biomechanics: Circulation; Springer: New York, NY, USA, 1997. [Google Scholar]
- Regmi, S.; Fu, A.; Luo, K.Q. High Shear Stresses under Exercise Condition Destroy Circulating Tumor Cells in a Microfluidic System. Sci. Rep. 2017, 7, 39975. [Google Scholar] [CrossRef] [Green Version]
- Follain, G.; Osmani, N.; Azevedo, S.; Allio, G.; Mercier, L.; Karreman, M.A.; Solecki, G.; Garcia-Leon, M.J.; Fekonja, N.; Chabannes, V.; et al. Hemodynamic forces tune the arrest, adhesion and extravasation of circulating tumor cells. Dev. Cell 2018, 45, 33–52. [Google Scholar] [CrossRef]
- Polacheck, W.J.; Kutys, M.L.; Yang, J.; Eyckmans, J.; Wu, Y.; Vasavada, H.; Hirschi, K.K.; Chen, C.S. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 2017, 552, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Wieland, E.; Rodriguez-Vita, J.; Liebler, S.S.; Mogler, C.; Moll, I.; Herberich, S.E.; Espinet, E.; Herpel, E.; Menuchin, A.; Chang-Claude, J.; et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017, 31, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Karreman, M.A.; Mercier, L.; Schieber, N.L.; Solecki, G.; Allio, G.; Winkler, F.; Ruthensteiner, B.; Goetz, J.G.; Schwab, Y. Fast and precise targeting of single tumor cells in vivo by multimodal correlative microscopy. J. Cell Sci. 2016, 129, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Im, J.H.; Fu, W.; Wang, H.; Bhatia, S.K.; Hammer, D.A.; Kowalska, M.A.; Muschel, R.J. Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res. 2004, 64, 8613–8619. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.A.; Asad, D.; Amu, E.; Hensley, M.T.; Cores, J.; Vandergriff, A.; Tang, J.; Dinh, P.U.; Shen, D.; Qiao, L.; et al. Circulating tumor cells exit circulation while maintaining multicellularity, augmenting metastatic potential. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Long, E.O. CD28 Homolog Is a Strong Activator of Natural Killer Cells for Lysis of B7H7(+) Tumor Cells. Cancer Immunol. Res. 2019, 7, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Rejniak, K.A. Circulating Tumor Cells: When a Solid Tumor Meets a Fluid Microenvironment. Adv. Exp. Med. Biol. 2016, 936, 93–106. [Google Scholar] [PubMed] [Green Version]
- Anderson, K.J.; de Guillebon, A.; Hughes, A.D.; Wang, W.; King, M.R. Effect of circulating tumor cell aggregate configuration on hemodynamic transport and wall contact. Math Biosci. 2017, 294, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Fang, F.; Zhang, Q. Circulating tumor cell clusters: What we know and what we expect (Review). Int. J. Oncol. 2016, 49, 2206–2216. [Google Scholar] [CrossRef] [Green Version]
- Mohammadalipour, A.; Burdick, M.M.; Tees, D.F.J. Deformability of breast cancer cells in correlation with surface markers and cell rolling. FASEB J. 2018, 32, 1806–1817. [Google Scholar] [CrossRef] [Green Version]
- Celia-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef] [Green Version]
- Janiszewska, M.; Tabassum, D.P.; Castano, Z.; Cristea, S.; Yamamoto, K.N.; Kingston, N.L.; Murphy, K.C.; Shu, S.; Harper, N.W.; Del Alcazar, C.G.; et al. Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nat. Cell Biol. 2019, 21, 879–888. [Google Scholar] [CrossRef]
- Burrell, R.A.; Swanton, C. Re-Evaluating Clonal Dominance in Cancer Evolution. Trends Cancer 2016, 2, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumor dormancy. Nat. Cell Biol. 2013, 15, 807. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Eyles, J.; Puaux, A.-L.; Wang, X.; Toh, B.; Prakash, C.; Hong, M.; Tan, T.G.; Zheng, L.; Ong, L.C.; Jin, Y.; et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Investig. 2010, 120, 2030–2039. [Google Scholar] [CrossRef]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massagué, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [Green Version]
- Castaño, Z.; San Juan, B.P.; Spiegel, A.; Pant, A.; DeCristo, M.J.; Laszewski, T.; Ubellacker, J.M.; Janssen, S.R.; Dongre, A.; Reinhardt, F.; et al. IL-1β inflammatory response driven by primary breast cancer prevents metastasis-initiating cell colonization. Nat. Cell Biol. 2018, 20, 1084–1097. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680. [Google Scholar] [CrossRef]
- Sceneay, J.; Chow, M.T.; Chen, A.; Halse, H.M.; Wong, C.S.F.; Andrews, D.M.; Sloan, E.K.; Parker, B.S.; Bowtell, D.D.; Smyth, M.J.; et al. Primary Tumor Hypoxia Recruits CD11b+/Ly6Cmed/Ly6G+ Immune Suppressor Cells and Compromises NK Cell Cytotoxicity in the Premetastatic Niche. Cancer Res. 2012, 72, 3906–3911. [Google Scholar] [CrossRef]
- Gil-Bernabe, A.M.; Ferjancic, S.; Tlalka, M.; Zhao, L.; Allen, P.D.; Im, J.H.; Watson, K.; Hill, S.A.; Amirkhosravi, A.; Francis, J.L.; et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 2012, 119, 3164–3175. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Najmeh, S.; Cools-Lartigue, J.; Rayes, R.F.; Gowing, S.; Vourtzoumis, P.; Bourdeau, F.; Giannias, B.; Berube, J.; Rousseau, S.; Ferri, L.E.; et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int. J. Cancer 2017, 140, 2321–2330. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Morel, A.-P.; Ginestier, C.; Pommier, R.M.; Cabaud, O.; Ruiz, E.; Wicinski, J.; Devouassoux-Shisheboran, M.; Combaret, V.; Finetti, P.; Chassot, C.; et al. A stemness-related ZEB1–MSRB3 axis governs cellular pliancy and breast cancer genome stability. Nat. Med. 2017, 23, 568–578. [Google Scholar] [CrossRef]
- Liu, Z.; Tu, K.; Wang, Y.; Yao, B.; Li, Q.; Wang, L.; Dou, C.; Liu, Q.; Zheng, X. Hypoxia Accelerates Aggressiveness of Hepatocellular Carcinoma Cells Involving Oxidative Stress, Epithelial-Mesenchymal Transition and Non-Canonical Hedgehog Signaling. Cell. Physiol. Biochem. 2017, 44, 1856–1868. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075. [Google Scholar] [CrossRef]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl. Acad. Sci. USA 2016, 113, E1555–E1564. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyenne, V.; Ghoroghi, S.; Collot, M.; Bons, J.; Follain, G.; Harlepp, S.; Mary, B.; Bauer, J.; Mercier, L.; Busnelli, I.; et al. Studying the Fate of Tumor Extracellular Vesicles at High Spatiotemporal Resolution Using the Zebrafish Embryo. Dev. Cell. 2019, 48, 554–572 e557. [Google Scholar] [CrossRef] [PubMed]
- Ombrato, L.; Nolan, E.; Kurelac, I.; Mavousian, A.; Bridgeman, V.L.; Heinze, I.; Chakravarty, P.; Horswell, S.; Gonzalez-Gualda, E.; Matacchione, G.; et al. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature 2019, 572, 603–608. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
San Juan, B.P.; Garcia-Leon, M.J.; Rangel, L.; Goetz, J.G.; Chaffer, C.L. The Complexities of Metastasis. Cancers 2019, 11, 1575. https://doi.org/10.3390/cancers11101575
San Juan BP, Garcia-Leon MJ, Rangel L, Goetz JG, Chaffer CL. The Complexities of Metastasis. Cancers. 2019; 11(10):1575. https://doi.org/10.3390/cancers11101575
Chicago/Turabian StyleSan Juan, Beatriz P., Maria J. Garcia-Leon, Laura Rangel, Jacky G. Goetz, and Christine L. Chaffer. 2019. "The Complexities of Metastasis" Cancers 11, no. 10: 1575. https://doi.org/10.3390/cancers11101575
APA StyleSan Juan, B. P., Garcia-Leon, M. J., Rangel, L., Goetz, J. G., & Chaffer, C. L. (2019). The Complexities of Metastasis. Cancers, 11(10), 1575. https://doi.org/10.3390/cancers11101575