Induction of NK Cell Reactivity against B-Cell Acute Lymphoblastic Leukemia by an Fc-Optimized FLT3 Antibody

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
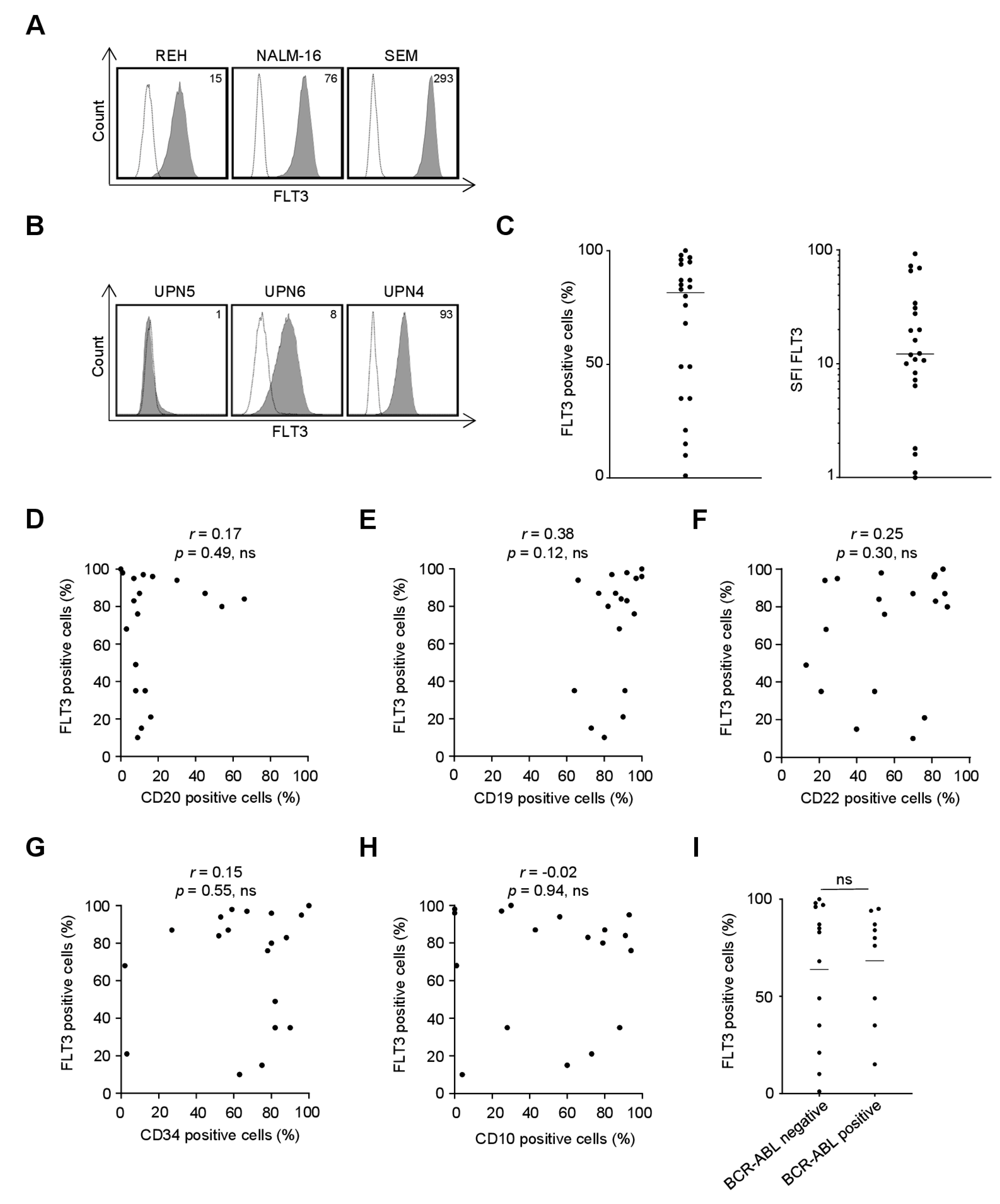
2.1. FLT3 Surface Expression on B-ALL Cell Lines and Primary Cells as Recognized by the FLT3 Binder 4G8
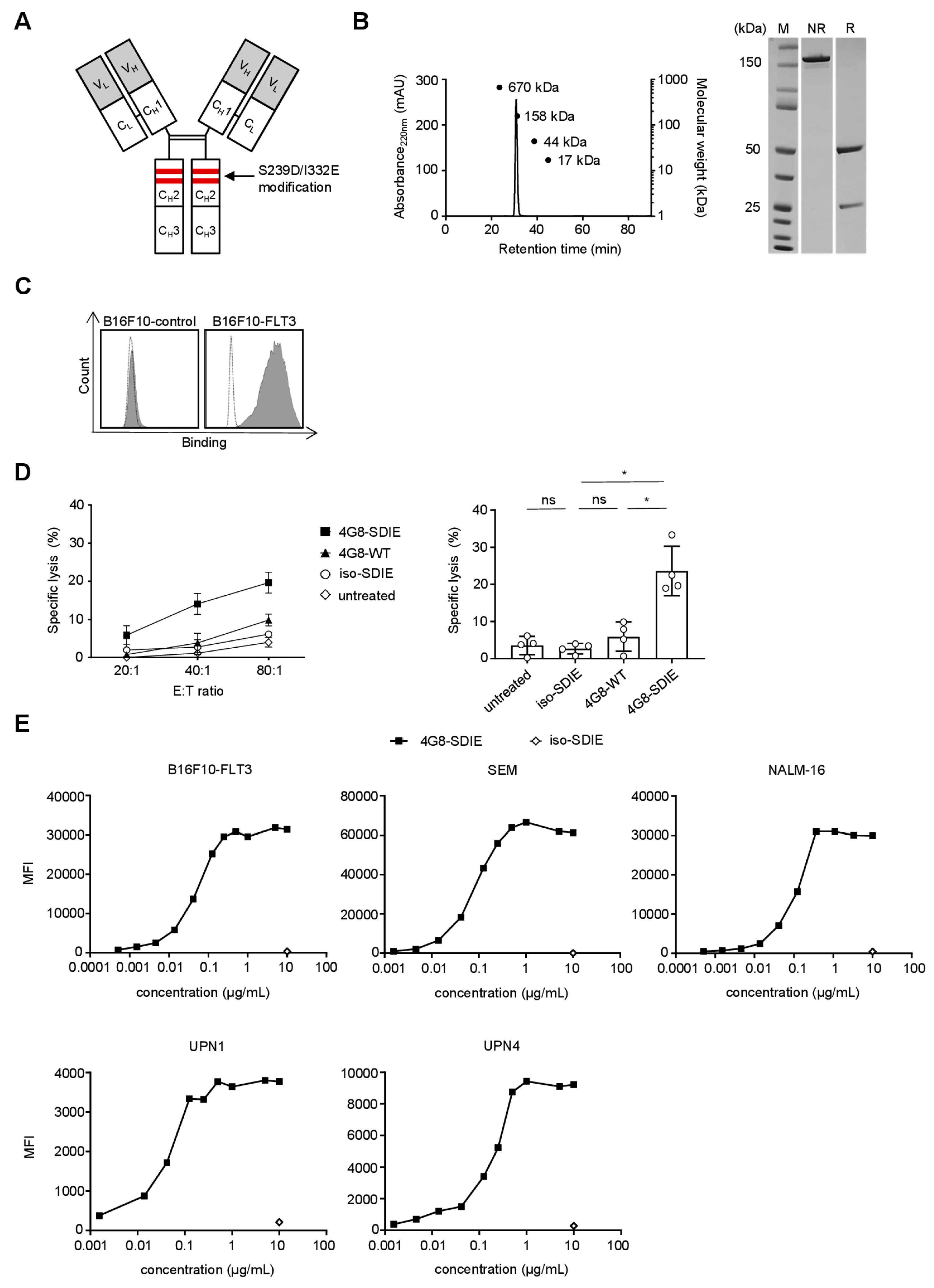
2.2. Production and Characterization of 4G8-SDIE
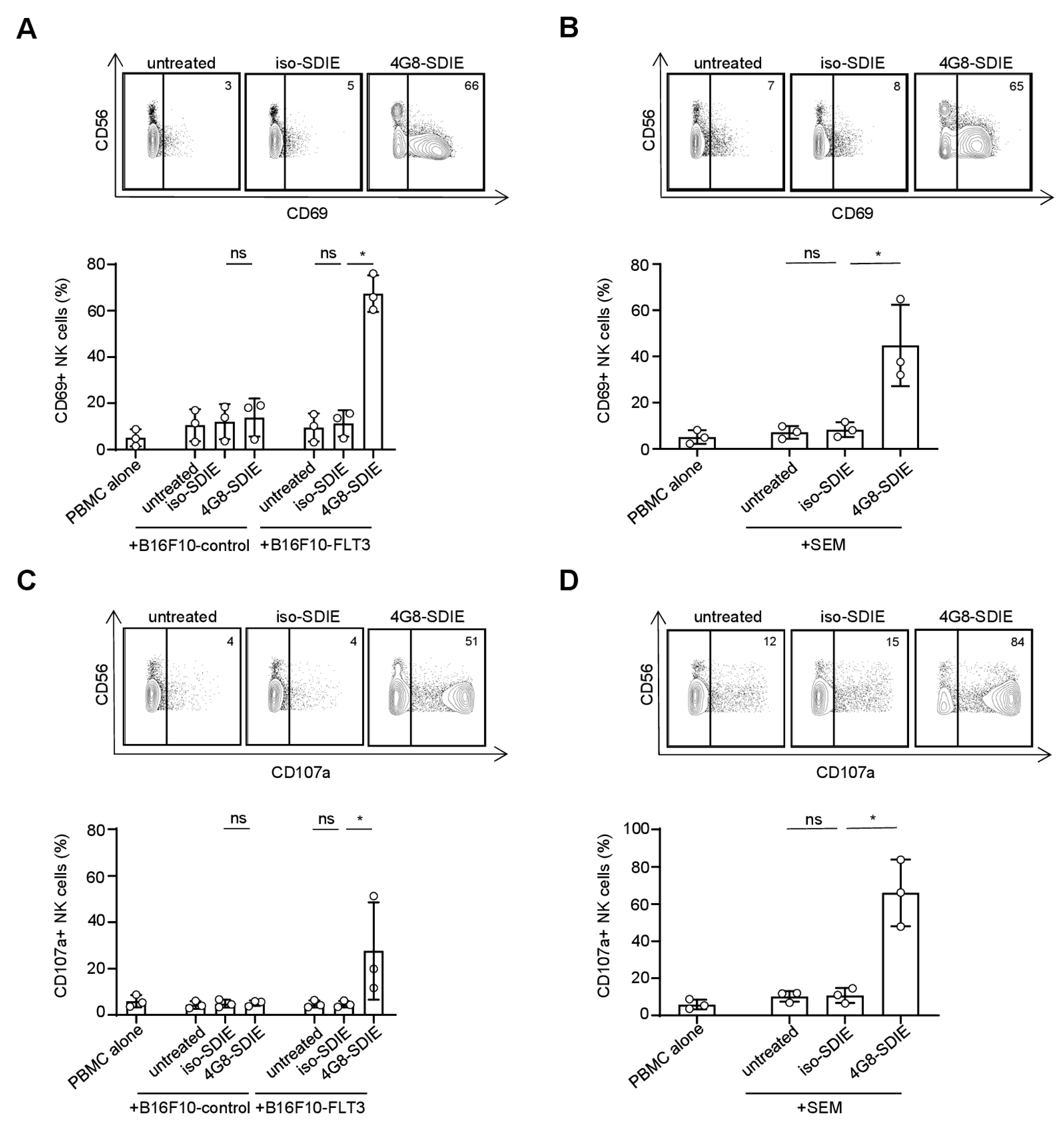
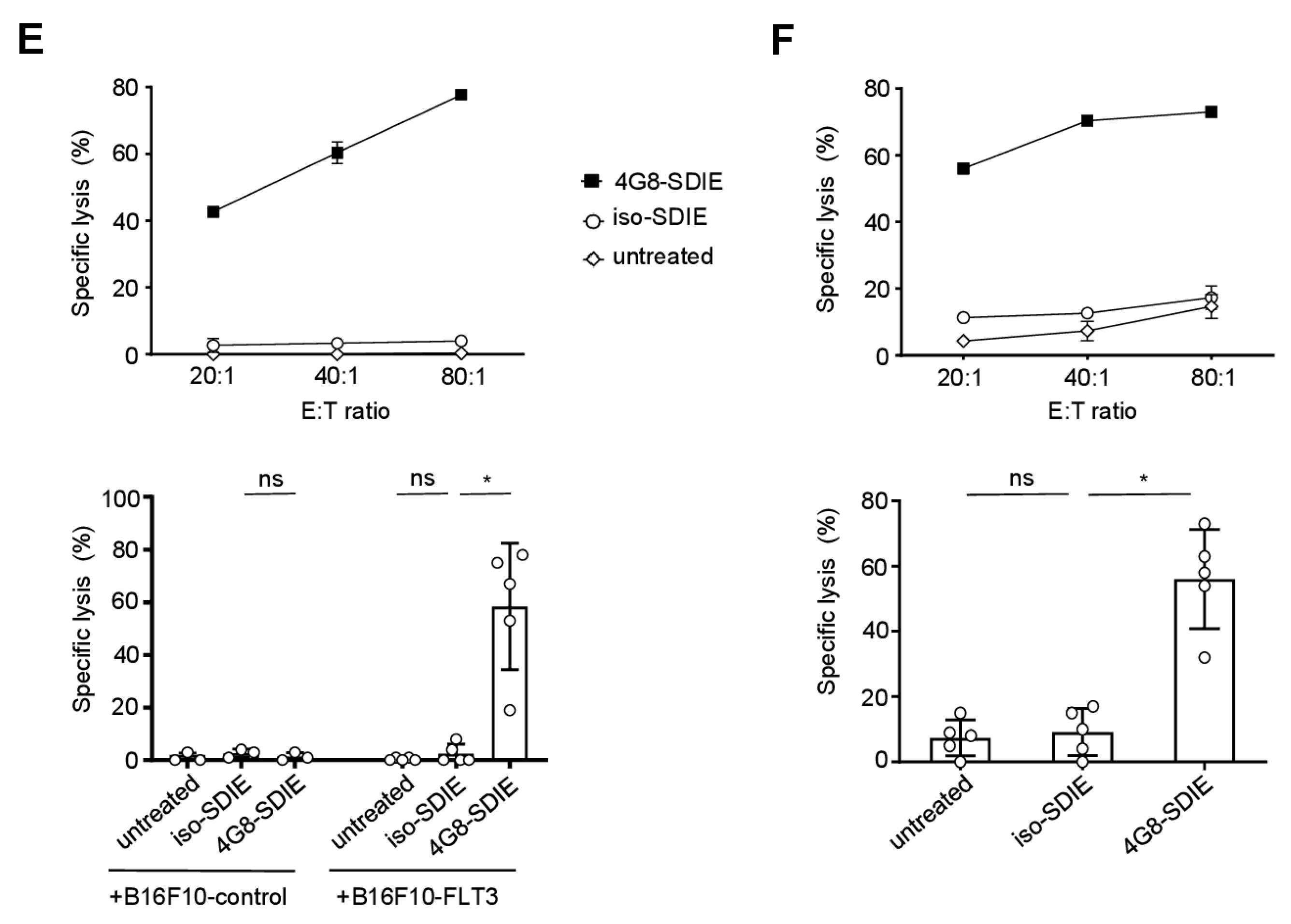
2.3. Induction of NK Cell Reactivity against FLT3+ Target Cells
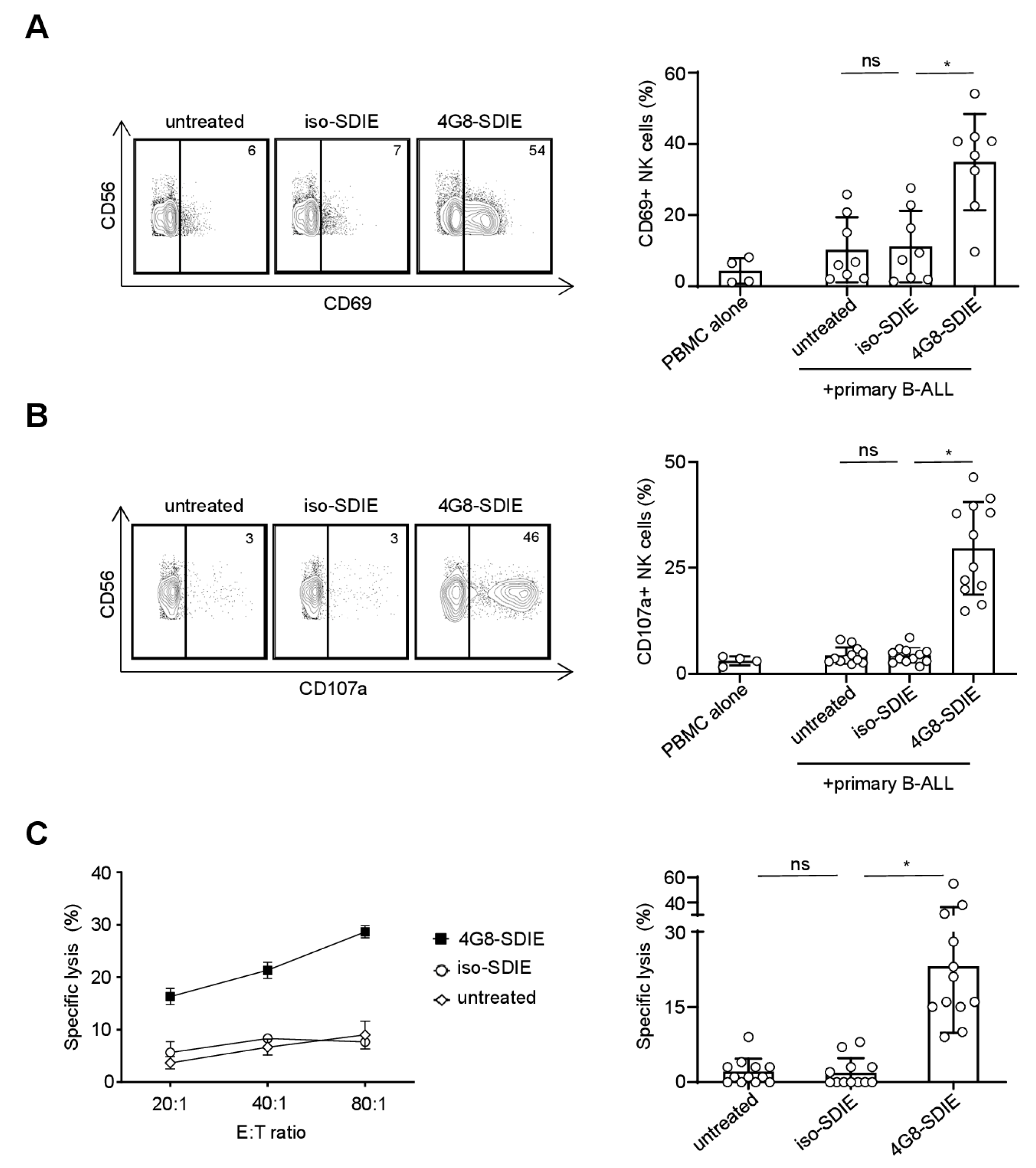
2.4. Induction of NK Cell Immunity Against Primary B-ALL Cells
3. Discussion
4. Materials and Methods
4.1. Production, Purification and Structural Analysis of Fc-Optimized Antibodies
4.2. Cells
4.3. Flow Cytometry
4.4. Analysis of NK Cell Activation and Degranulation
4.5. Analysis of NK Cell Cytotoxicity
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rothschilds, A.M.; Wittrup, K.D. What, Why, Where, and When: Bringing Timing to Immuno-Oncology. Trends Immunol. 2019, 40, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2012, 9, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Rituximab: A review of its use in chronic lymphocytic leukaemia, low-grade or follicular lymphoma and diffuse large B-cell lymphoma. Drugs 2010, 70, 1445–1476. [Google Scholar] [CrossRef] [PubMed]
- Kellner, C.; Otte, A.; Cappuzzello, E.; Klausz, K.; Peipp, M. Modulating Cytotoxic Effector Functions by Fc Engineering to Improve Cancer Therapy. Transfus. Med. Hemother. 2017, 44, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Seidel, U.J.; Schlegel, P.; Lang, P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 2013, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gokbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Grosse-Hovest, L.; Nubling, T.; Pyz, E.; Bamberg, M.L.; Aulwurm, S.; Buhring, H.J.; Schwartz, K.; Haen, S.P.; Schilbach, K.; et al. Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 2012, 26, 1228–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmiedel, B.J.; Werner, A.; Steinbacher, J.; Nuebling, T.; Buechele, C.; Grosse-Hovest, L.; Salih, H.R. Generation and Preclinical Characterization of a Fc-optimized GITR-Ig Fusion Protein for Induction of NK Cell Reactivity Against Leukemia. Mol. Ther. 2013, 21, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmiedel, B.J.; Scheible, C.A.; Nuebling, T.; Kopp, H.G.; Wirths, S.; Azuma, M.; Schneider, P.; Jung, G.; Grosse-Hovest, L.; Salih, H.R. RANKL Expression, Function, and Therapeutic Targeting in Multiple Myeloma and Chronic Lymphocytic Leukemia. Cancer Res. 2013, 73, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbacher, J.; Baltz-Ghahremanpour, K.; Schmiedel, B.J.; Steinle, A.; Jung, G.; Kubler, A.; Andre, M.C.; Grosse-Hovest, L.; Salih, H.R. An Fc-optimized NKG2D-immunoglobulin G fusion protein for induction of natural killer cell reactivity against leukemia. Int. J. Cancer 2015, 136, 1073–1084. [Google Scholar] [CrossRef]
- Koerner, S.P.; Andre, M.C.; Leibold, J.S.; Kousis, P.C.; Kubler, A.; Pal, M.; Haen, S.P.; Buhring, H.J.; Grosse-Hovest, L.; Jung, G.; et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leukemia 2017, 31, 459–469. [Google Scholar] [CrossRef]
- Raab, S.; Steinbacher, J.; Schmiedel, B.J.; Kousis, P.C.; Steinle, A.; Jung, G.; Grosse-Hovest, L.; Salih, H.R. Fc-optimized NKG2D-Fc constructs induce NK cell antibody-dependent cellular cytotoxicity against breast cancer cells independently of HER2/neu expression status. J. Immunol. 2014, 193, 4261–4272. [Google Scholar] [CrossRef] [Green Version]
- Schmied, B.J.; Riegg, F.; Zekri, L.; Grosse-Hovest, L.; Buhring, H.J.; Jung, G.; Salih, H.R. An Fc-Optimized CD133 Antibody for Induction of Natural Killer Cell Reactivity against Colorectal Cancer. Cancers (Basel) 2019, 11, 789. [Google Scholar] [CrossRef] [Green Version]
- Drexler, H.G. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia 1996, 10, 588–599. [Google Scholar]
- Raponi, S.; De Propris, M.S.; Intoppa, S.; Milani, M.L.; Vitale, A.; Elia, L.; Perbellini, O.; Pizzolo, G.; Foa, R.; Guarini, A. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: Analysis of 552 cases. Leuk. Lymphoma 2011, 52, 1098–1107. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Kantarjian, H.; Jabbour, E. Recent Advances in Adult Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2019, 14, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Hefazi, M.; Litzow, M.R. Recent advances in the biology and treatment of B-cell acute lymphoblastic leukemia. Blood Lymphat. Cancer 2018, 8, 47–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinner, S.; Liedtke, M. Antibody-based therapies in patients with acute lymphoblastic leukemia. Hematology 2018, 2018, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellner, C.; Peipp, M.; Gramatzki, M.; Schrappe, M.; Schewe, D.M. Perspectives of Fc engineered antibodies in CD19 targeting immunotherapies in pediatric B-cell precursor acute lymphoblastic leukemia. Oncoimmunology 2018, 7, e1448331. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Paul, M.R.; Wong, V.; Aristizabal, P.; Kuo, D.J. Treatment of Recurrent Refractory Pediatric Pre-B Acute Lymphoblastic Leukemia Using Inotuzumab Ozogamicin Monotherapy Resulting in CD22 Antigen Expression Loss as a Mechanism of Therapy Resistance. J. Pediatr. Hematol. Oncol. 2019, 41, e546–549. [Google Scholar] [CrossRef]
- Wynne, J.; Wright, D.; Stock, W. Inotuzumab: From preclinical development to success in B-cell acute lymphoblastic leukemia. Blood Adv. 2019, 3, 96–104. [Google Scholar] [CrossRef]
- Bhojwani, D.; Sposto, R.; Shah, N.N.; Rodriguez, V.; Yuan, C.; Stetler-Stevenson, M.; O’Brien, M.M.; McNeer, J.L.; Quereshi, A.; Cabannes, A.; et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. Leukemia 2019, 33, 884–892. [Google Scholar] [CrossRef]
- Liu, D.; Zhao, J.; Song, Y.; Luo, X.; Yang, T. Clinical trial update on bispecific antibodies, antibody-drug conjugates, and antibody-containing regimens for acute lymphoblastic leukemia. J. Hematol. Oncol. 2019, 12, 15. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.S.; Illmer, T.; et al. Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, A.; Ayello, J.; Van de Ven, C.; Elmacken, M.; Sabulski, A.; Barth, M.J.; Czuczman, M.S.; Islam, H.; Klein, C.; Cairo, M.S. Obinutuzumab (GA101) compared to rituximab significantly enhances cell death and antibody-dependent cytotoxicity and improves overall survival against CD20+ rituximab-sensitive/-resistant Burkitt lymphoma (BL) and precursor B-acute lymphoblastic leukaemia (pre-B-ALL): Potential targeted therapy in patients with poor risk CD20+ BL and pre-B-ALL. Br. J. Haematol. 2015, 171, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Blatt, K.; Menzl, I.; Eisenwort, G.; Cerny-Reiterer, S.; Herrmann, H.; Herndlhofer, S.; Stefanzl, G.; Sadovnik, I.; Berger, D.; Keller, A.; et al. Phenotyping and Target Expression Profiling of CD34+/CD38− and CD34+/CD38+ Stem- and Progenitor cells in Acute Lymphoblastic Leukemia. Neoplasia 2018, 20, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Chougule, R.A.; Shah, K.; Moharram, S.A.; Vallon-Christersson, J.; Kazi, J.U. Glucocorticoid-resistant B cell acute lymphoblastic leukemia displays receptor tyrosine kinase activation. NPJ Genom. Med. 2019, 4, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vora, H.H.; Shukla, S.N.; Brahambhatt, B.V.; Mehta, S.H.; Patel, N.A.; Parikh, S.K.; Shah, K.N.; Shah, P.M. Clinical relevance of FLT3 receptor protein expression in Indian patients with acute leukemia. Asia Pac. J. Clin. Oncol. 2010, 6, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.W.; Schneider, P.; de Lorenzo, P.; Valsecchi, M.G.; den Boer, M.L.; Pieters, R. Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute lymphoblastic leukemia. Blood 2007, 110, 2774–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chillon, M.C.; Gomez-Casares, M.T.; Lopez-Jorge, C.E.; Rodriguez-Medina, C.; Molines, A.; Sarasquete, M.E.; Alcoceba, M.; Miguel, J.D.; Bueno, C.; Montes, R.; et al. Prognostic significance of FLT3 mutational status and expression levels in MLL-AF4+ and MLL-germline acute lymphoblastic leukemia. Leukemia 2012, 26, 2360–2366. [Google Scholar] [CrossRef]
- Oelsner, S.; Waldmann, A.; Billmeier, A.; Roder, J.; Lindner, A.; Ullrich, E.; Marschalek, R.; Dotti, G.; Jung, G.; Grosse-Hovest, L.; et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int. J. Cancer 2019, 145, 1935–1945. [Google Scholar] [CrossRef]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3−ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef]
- Durben, M.; Schmiedel, D.; Hofmann, M.; Vogt, F.; Nubling, T.; Pyz, E.; Buhring, H.J.; Rammensee, H.G.; Salih, H.R.; Grosse-Hovest, L.; et al. Characterization of a bispecific FLT3 X CD3 antibody in an improved, recombinant format for the treatment of leukemia. Mol. Ther. 2015, 23, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Tsuzuki, S.; Akahori, Y.; Ukai, Y.; Sumitomo, M.; Murayama, Y.; Yamamoto, K.; Inaguma, Y.; Tokuda, M.; Abe, A.; et al. Isolation of human mAbs that directly modulate FMS-related tyrosine kinase 3 signaling. Cancer Sci. 2012, 103, 350–359. [Google Scholar] [CrossRef]
- Piloto, O.; Nguyen, B.; Huso, D.; Kim, K.T.; Li, Y.W.; Witte, L.; Hicklin, D.J.; Brown, P.; Small, D. IMC-EB10, an anti-FLT3 monoclonal antibody, prolongs survival and reduces nonobese diabetic/severe combined immunodeficient engraftment of some acute lymphoblastic leukemia cell lines and primary leukemic samples. Cancer Res. 2006, 66, 4843–4851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörfel, D.; Döhner, K.; Kapp-Schwoerer, S.; Kayser, S.; Thol, F.; Heuser, M.; Grosse-Hovest, L.; Kanz, L.; Schlenk, R.F.; Jung, G.; et al. A first in man study with a Fc-optimized FLT3 antibody for treatment of acute myeloid leukemia with minimal residual disease. Oncol. Res. Treat. 2018, 41 (Suppl. 4), 1–358. [Google Scholar] [CrossRef]
- Goyon, A.; D’Atri, V.; Colas, O.; Fekete, S.; Beck, A.; Guillarme, D. Characterization of 30 therapeutic antibodies and related products by size exclusion chromatography: Feasibility assessment for future mass spectrometry hyphenation. J. Chromatogr. B 2017, 1065, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S. Effects of protein aggregates: An immunologic perspective. AAPS J. 2006, 8, E501–E507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, M.K.; Deshpande, M.; Yang, J.; Reynolds, H.; Bryson, C.; Fogg, M.; Baker, M.P.; Herskovitz, J.; Goletz, T.J.; Zhou, L.; et al. Use of In Vitro Assays to Assess Immunogenicity Risk of Antibody-Based Biotherapeutics. PLoS ONE 2016, 11, e0159328. [Google Scholar] [CrossRef]
- Sawalha, Y.; Advani, A.S. Management of older adults with acute lymphoblastic leukemia: Challenges & current approaches. Int. J. Hematol. Oncol. 2018, 7, IJH02. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmied, B.J.; Lutz, M.S.; Riegg, F.; Zekri, L.; Heitmann, J.S.; Bühring, H.-J.; Jung, G.; Salih, H.R. Induction of NK Cell Reactivity against B-Cell Acute Lymphoblastic Leukemia by an Fc-Optimized FLT3 Antibody. Cancers 2019, 11, 1966. https://doi.org/10.3390/cancers11121966
Schmied BJ, Lutz MS, Riegg F, Zekri L, Heitmann JS, Bühring H-J, Jung G, Salih HR. Induction of NK Cell Reactivity against B-Cell Acute Lymphoblastic Leukemia by an Fc-Optimized FLT3 Antibody. Cancers. 2019; 11(12):1966. https://doi.org/10.3390/cancers11121966
Chicago/Turabian StyleSchmied, Bastian J., Martina S. Lutz, Fabian Riegg, Latifa Zekri, Jonas S. Heitmann, Hans-Jörg Bühring, Gundram Jung, and Helmut R. Salih. 2019. "Induction of NK Cell Reactivity against B-Cell Acute Lymphoblastic Leukemia by an Fc-Optimized FLT3 Antibody" Cancers 11, no. 12: 1966. https://doi.org/10.3390/cancers11121966
APA StyleSchmied, B. J., Lutz, M. S., Riegg, F., Zekri, L., Heitmann, J. S., Bühring, H. -J., Jung, G., & Salih, H. R. (2019). Induction of NK Cell Reactivity against B-Cell Acute Lymphoblastic Leukemia by an Fc-Optimized FLT3 Antibody. Cancers, 11(12), 1966. https://doi.org/10.3390/cancers11121966