Cell-Free DNA Variant Sequencing Using CTC-Depleted Blood for Comprehensive Liquid Biopsy Testing in Metastatic Breast Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
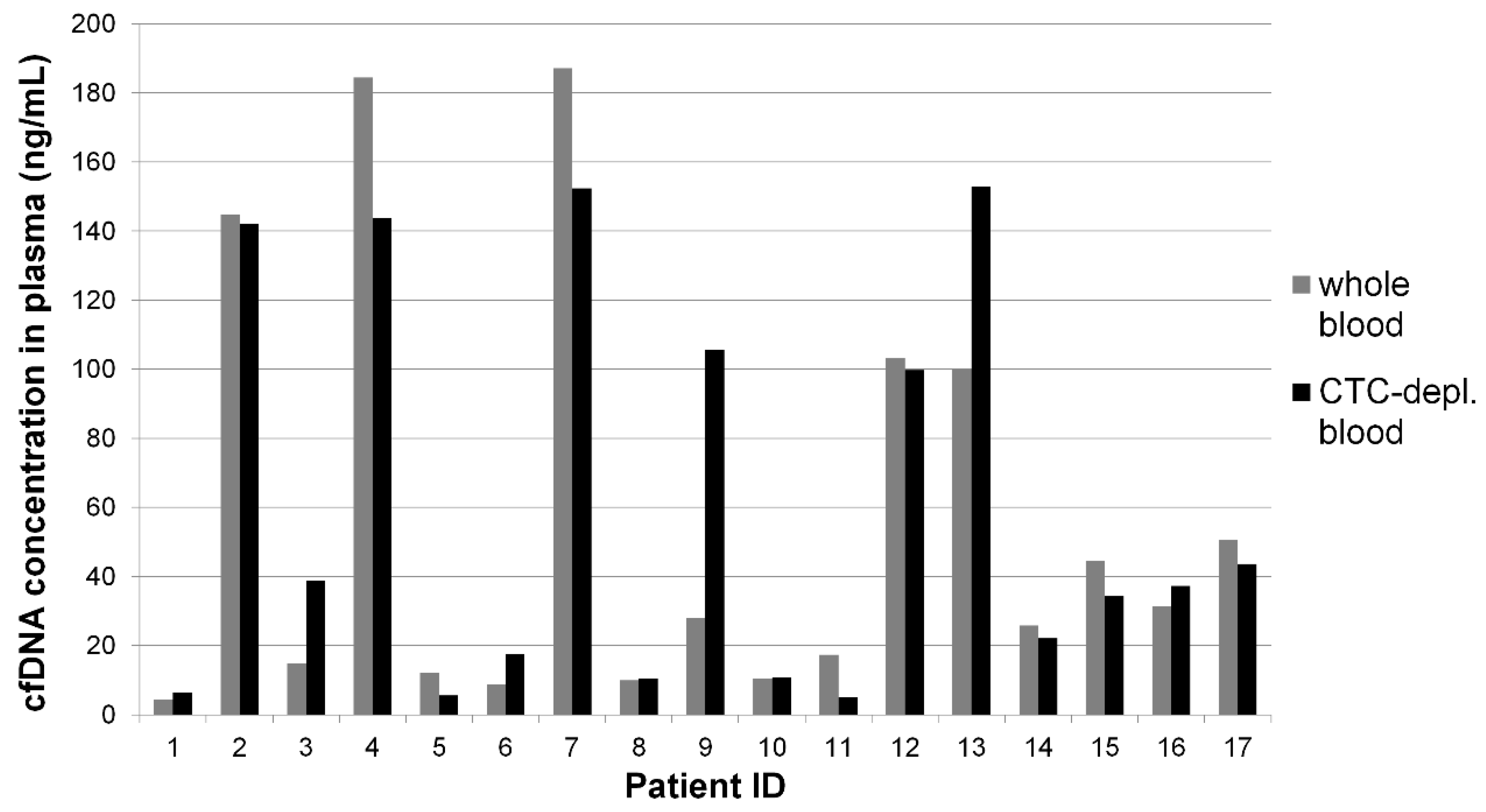
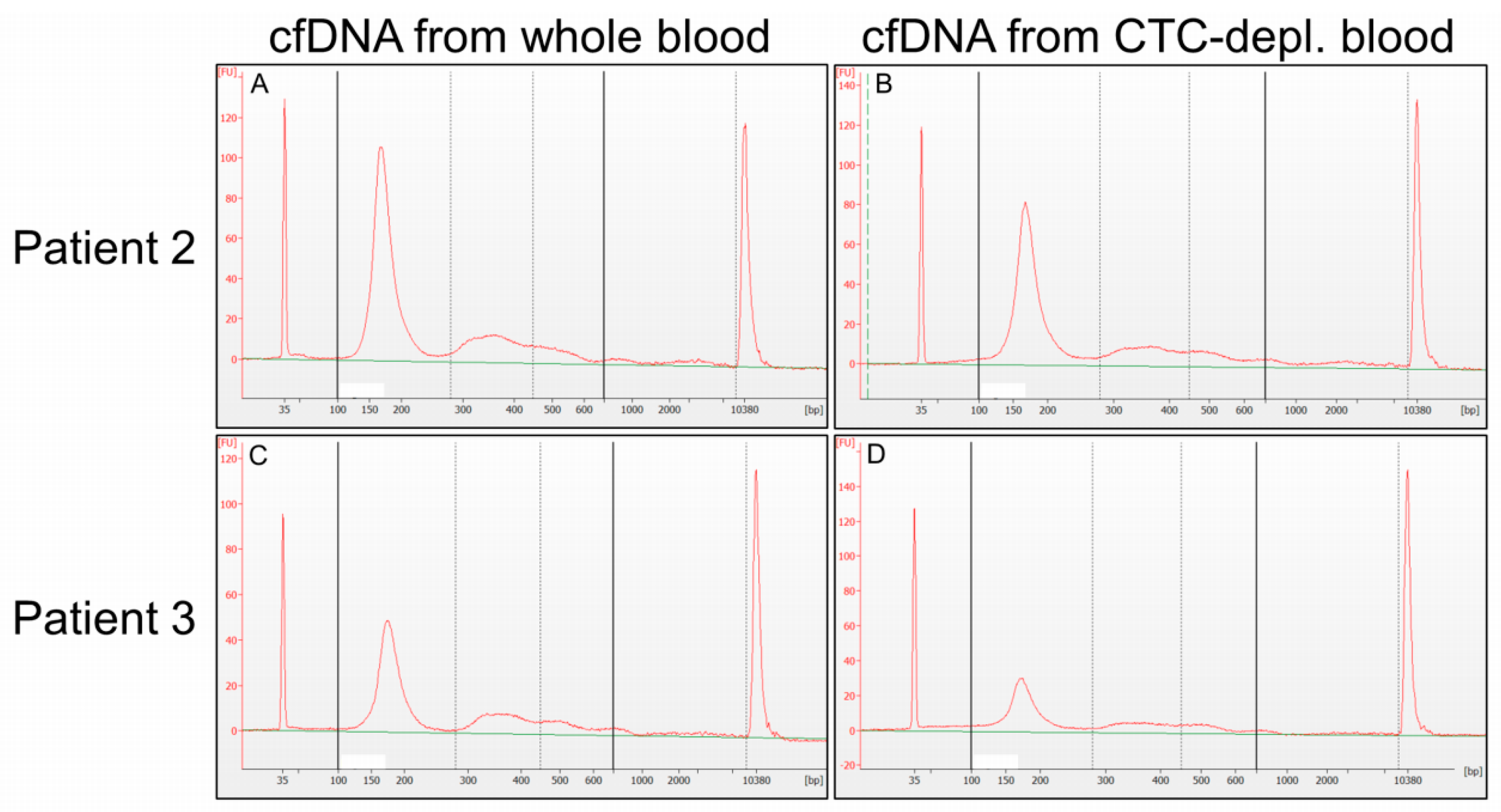
2.2. Cell-Free DNA Concentration and Fragmentation
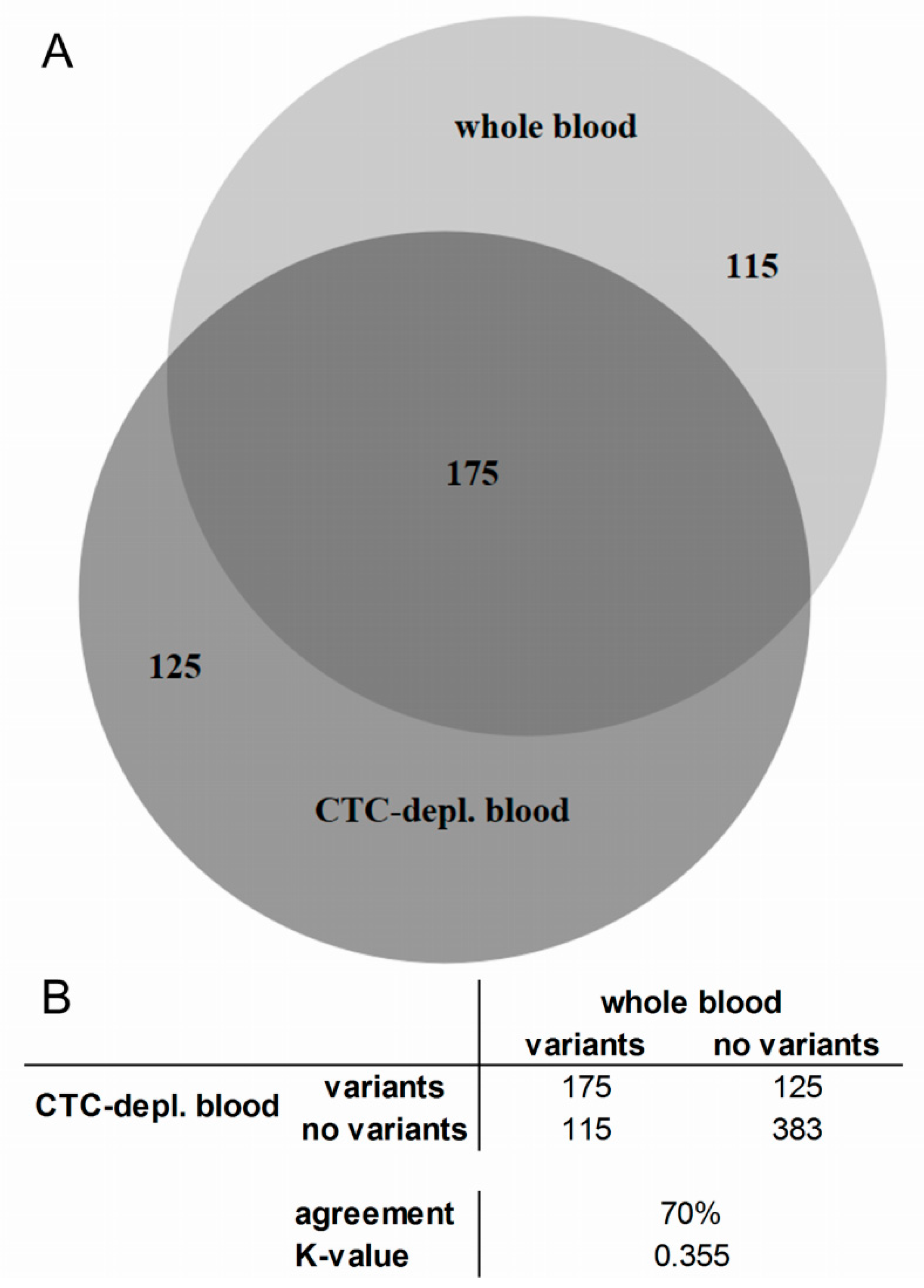
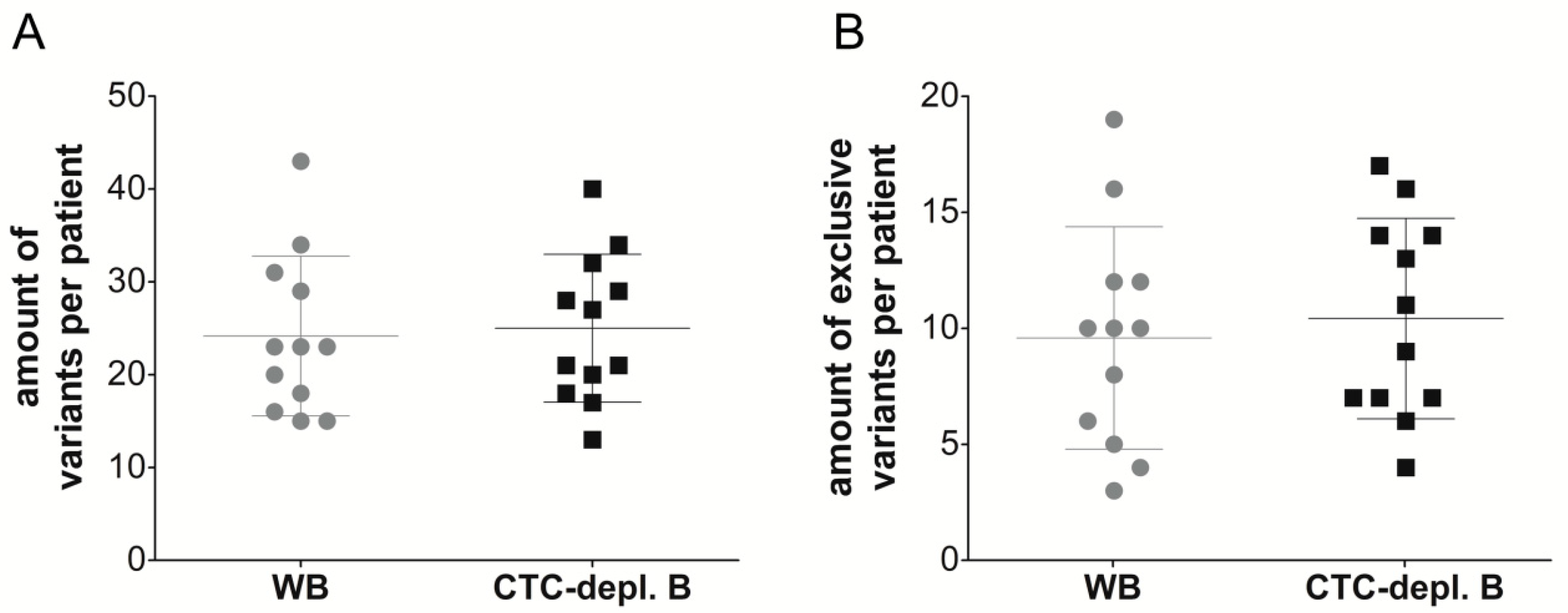
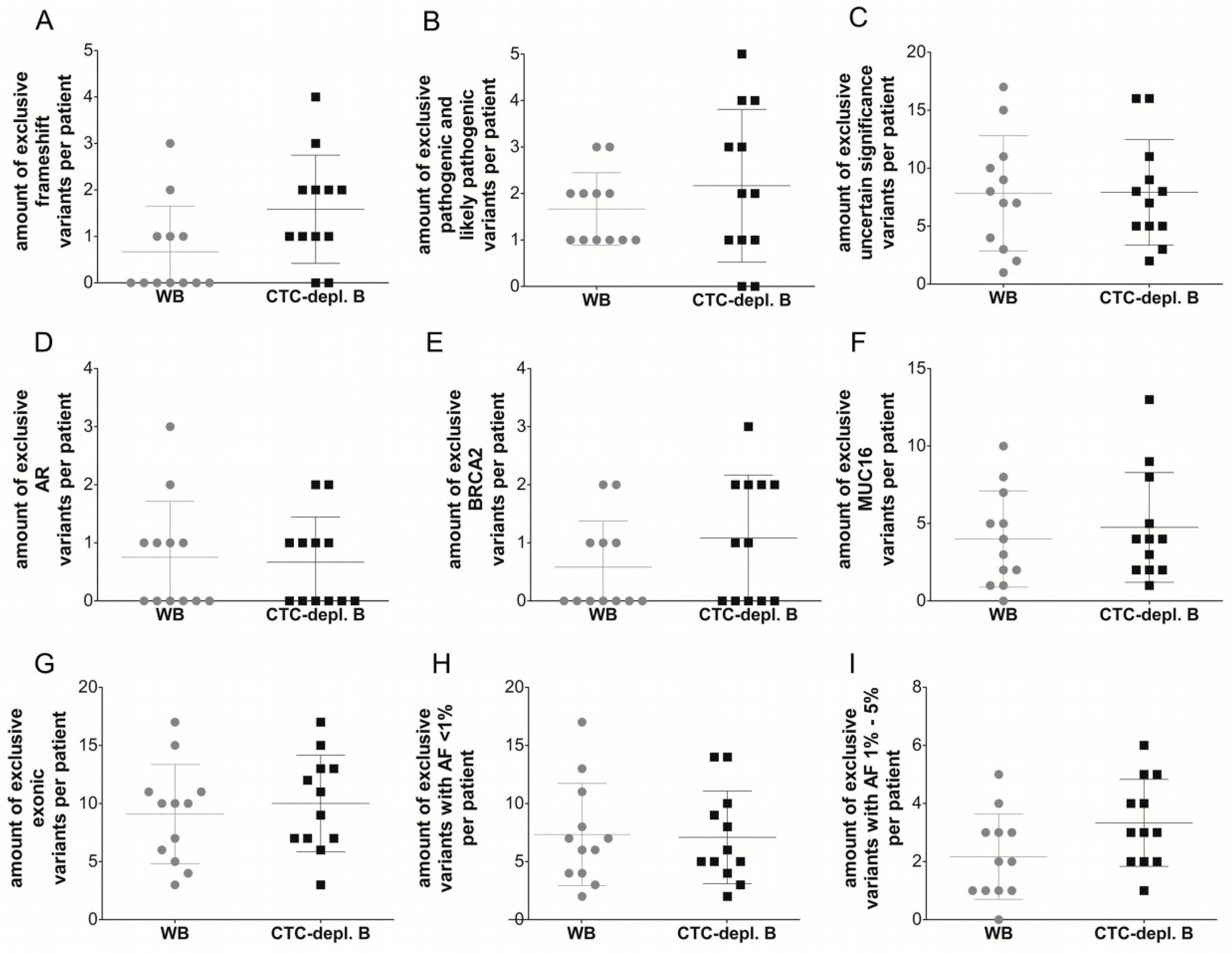
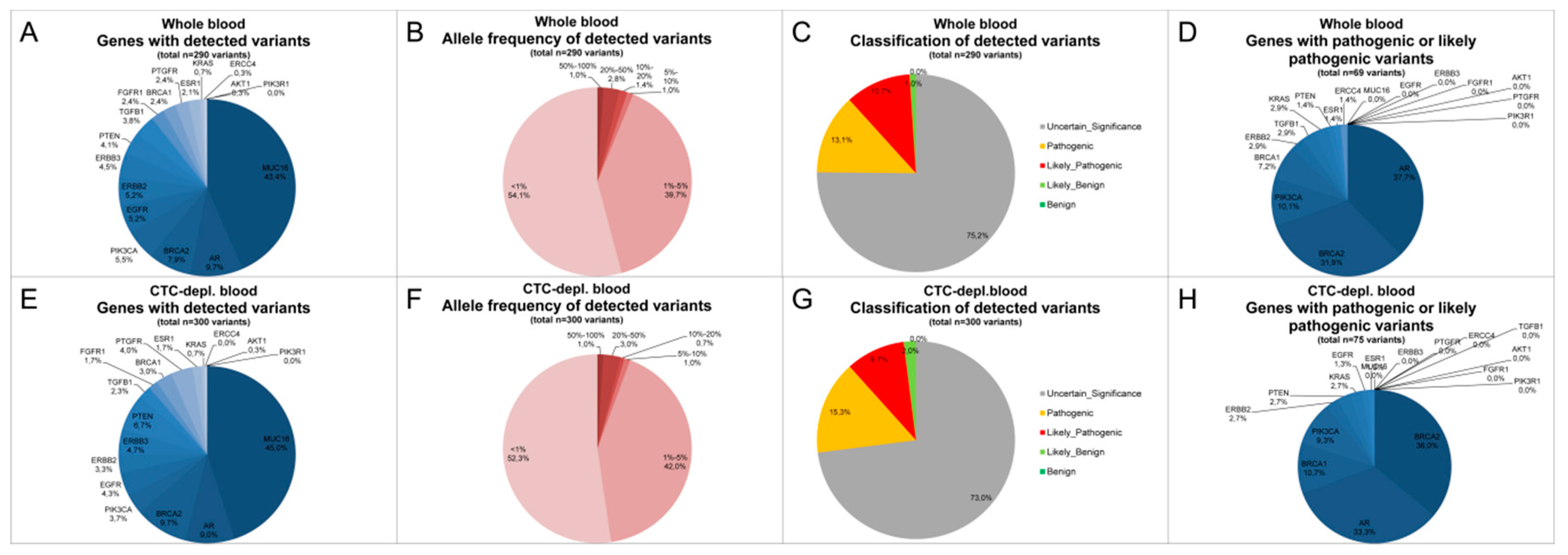
2.3. Cell-Free DNA Variants from Matched Whole Blood and Circulating Tumor Cell-Depleted Blood
2.4. Clinically Relevant Characteristics of Cell-Free DNA Variants and Circulating Tumor Cell Expression
3. Discussion
3.1. Cell-Free DNA Sequencing from Circulating Tumor Cell-Depleted Blood Favored in the Future
3.2. Additive Value of Cell-Free DNA Mutational and Circulating Tumor Cell Transcriptional Analyses
4. Materials and Methods
4.1. Patient Population Characteristics and Eligibility Criteria
4.2. Sampling of Blood, Isolation of Circulating Tumor Cells and Processing of Plasma
4.3. Isolation of Cell-Free DNA
4.4. Cell-Free DNA Quantification
4.5. Library Construction
4.6. Sequencing
4.7. Data Analysis/Bioinformatical Analysis
4.8. Messenger RNA Isolation and Quantitative PCR
4.9. Statistical analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diaz, L.A.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, N.; Fehm, T.; Banys-Paluchowski, M.; Janni, W.; Schramm, A. Liquid Biopsy in Metastasized Breast Cancer as Basis for Treatment Decisions. Oncol. Res. Treat. 2016, 39, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, T.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Tomiguchi, M.; Sueta, A.; Murakami, K.; Omoto, Y.; Iwase, H. Analysis of ESR1 and PIK3CA mutations in plasma cell-free DNA from ER-positive breast cancer patients. Oncotarget 2017, 8, 52142–52155. [Google Scholar] [CrossRef] [PubMed]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.-J.; Tsui, D.W.Y.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.-F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.M.M.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef]
- Hayes, D.F.; Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Miller, M.C.; Matera, J.; Allard, W.J.; Doyle, G.V.; Terstappen, L.W.W.M. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin. Cancer Res. 2006, 12, 4218–4224. [Google Scholar] [CrossRef]
- Budd, G.T.; Cristofanilli, M.; Ellis, M.J.; Stopeck, A.; Borden, E.; Miller, M.C.; Matera, J.; Repollet, M.; Doyle, G.V.; Terstappen, L.W.M.M.; et al. Circulating tumor cells versus imaging—Predicting overall survival in metastatic breast cancer. Clin. Cancer Res. 2006, 12, 6403–6409. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Circulating tumor cells: Liquid biopsy of cancer. Clin. Chem. 2013, 59, 110–118. [Google Scholar] [CrossRef]
- Keup, C.; Mach, P.; Aktas, B.; Tewes, M.; Kolberg, H.-C.; Hauch, S.; Sprenger-Haussels, M.; Kimmig, R.; Kasimir-Bauer, S. RNA Profiles of Circulating Tumor Cells and Extracellular Vesicles for Therapy Stratification of Metastatic Breast Cancer Patients. Clin. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat. Commun. 2018, 9, 1691. [Google Scholar] [CrossRef] [PubMed]
- Haber, D.A.; Velculescu, V.E. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef]
- Heidary, M.; Auer, M.; Ulz, P.; Heitzer, E.; Petru, E.; Gasch, C.; Riethdorf, S.; Mauermann, O.; Lafer, I.; Pristauz, G.; et al. The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Res. 2014, 16, 421. [Google Scholar] [CrossRef] [PubMed]
- Delfau-Larue, M.-H.; van der Gucht, A.; Dupuis, J.; Jais, J.-P.; Nel, I.; Beldi-Ferchiou, A.; Hamdane, S.; Benmaad, I.; Laboure, G.; Verret, B.; et al. Total metabolic tumor volume, circulating tumor cells, cell-free DNA: Distinct prognostic value in follicular lymphoma. Blood Adv. 2018, 2, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.A.; Guttery, D.S.; Hills, A.; Fernandez-Garcia, D.; Page, K.; Rosales, B.M.; Goddard, K.S.; Hastings, R.K.; Luo, J.; Ogle, O.; et al. Mutation Analysis of Cell-Free DNA and Single Circulating Tumor Cells in Metastatic Breast Cancer Patients with High Circulating Tumor Cell Counts. Clin. Cancer Res. 2017, 23, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Mu, Z.; Rademaker, A.W.; Austin, L.K.; Strickland, K.S.; Costa, R.L.B.; Nagy, R.J.; Zagonel, V.; Taxter, T.J.; Behdad, A.; et al. Cell-Free DNA and Circulating Tumor Cells: Comprehensive Liquid Biopsy Analysis in Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 560–568. [Google Scholar] [CrossRef]
- Mastoraki, S.; Strati, A.; Tzanikou, E.; Chimonidou, M.; Politaki, E.; Voutsina, A.; Psyrri, A.; Georgoulias, V.; Lianidou, E. ESR1 Methylation: A Liquid Biopsy-Based Epigenetic Assay for the Follow-up of Patients with Metastatic Breast Cancer Receiving Endocrine Treatment. Clin. Cancer Res. 2018, 24, 1500–1510. [Google Scholar] [CrossRef]
- Madhavan, D.; Wallwiener, M.; Bents, K.; Zucknick, M.; Nees, J.; Schott, S.; Cuk, K.; Riethdorf, S.; Trumpp, A.; Pantel, K.; et al. Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res. Treat. 2014, 146, 163–174. [Google Scholar] [CrossRef]
- Coco, S.; Alama, A.; Vanni, I.; Fontana, V.; Genova, C.; Dal Bello, M.G.; Truini, A.; Rijavec, E.; Biello, F.; Sini, C.; et al. Circulating Cell-Free DNA and Circulating Tumor Cells as Prognostic and Predictive Biomarkers in Advanced Non-Small Cell Lung Cancer Patients Treated with First-Line Chemotherapy. Int. J. Mol. Sci. 2017, 18, 1035. [Google Scholar] [CrossRef] [PubMed]
- Vo, J.H.; Nei, W.L.; Hu, M.; Phyo, W.M.; Wang, F.; Fong, K.W.; Tan, T.; Soong, Y.L.; Cheah, S.L.; Sommat, K.; et al. Comparison of Circulating Tumour Cells and Circulating Cell-Free Epstein-Barr Virus DNA in Patients with Nasopharyngeal Carcinoma Undergoing Radiotherapy. Sci. Rep. 2016, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Chimonidou, M.; Strati, A.; Malamos, N.; Kouneli, S.; Georgoulias, V.; Lianidou, E. Direct comparison study of DNA methylation markers in EpCAM-positive circulating tumour cells, corresponding circulating tumour DNA, and paired primary tumours in breast cancer. Oncotarget 2017, 8, 72054–72068. [Google Scholar] [CrossRef] [PubMed]
- Chimonidou, M.; Strati, A.; Malamos, N.; Georgoulias, V.; Lianidou, E.S. SOX17 promoter methylation in circulating tumor cells and matched cell-free DNA isolated from plasma of patients with breast cancer. Clin. Chem. 2013, 59, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Keup, C.; Hahn, P.; Hauch, S.; Sprenger-Haussels, M.; Tewes, M.; Mach, P.; Bittner, A.-K.; Kimmig, R.; Kasimir-Bauer, S.; Benyaa, K. Targeted PCR-based deep sequencing of cfDNA with unique molecular indices by a customized QIAseq Targeted DNA Panel. Protocols 2018. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [PubMed]
- Keup, C.; Benyaa, K.; Hauch, S.; Sprenger-Haussels, M.; Tewes, M.; Mach, P.; Bittner, A.-K.; Kimmig, R.; Hahn, P.; Kasimir-Bauer, S. Variants in cell-free DNA of hormone receptor positive, HER2 negative metastatic breast cancer assessed by targeted deep sequencing. Cell. Mol. Life Sci. 2018. submitted. [Google Scholar]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men with Metastatic Castration-Resistant Prostate Cancer Treated with First- and Second-Line Abiraterone and Enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gómez, H.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014, 16, 406. [Google Scholar] [CrossRef] [PubMed]
- Rahim, B.; O’Regan, R. AR Signaling in Breast Cancer. Cancers 2017, 9, 21. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.S.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Arch. Pathol. Lab. Med. 2014, 138, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Aktas, B.; Kasimir-Bauer, S.; Müller, V.; Janni, W.; Fehm, T.; Wallwiener, D.; Pantel, K.; Tewes, M. Comparison of the HER2, estrogen and progesterone receptor expression profile of primary tumor, metastases and circulating tumor cells in metastatic breast cancer patients. BMC Cancer 2016, 16, 522. [Google Scholar] [CrossRef] [PubMed]
- Lianidou, E.; Pantel, K. Liquid Biopsies. Genes Chromosomes Cancer 2019, 58, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Bulfoni, M.; Gerratana, L.; Del Ben, F.; Marzinotto, S.; Sorrentino, M.; Turetta, M.; Scoles, G.; Toffoletto, B.; Isola, M.; Beltrami, C.A.; et al. In patients with metastatic breast cancer the identification of circulating tumor cells in epithelial-to-mesenchymal transition is associated with a poor prognosis. Breast Cancer Res. 2016, 18, 30. [Google Scholar] [CrossRef] [PubMed]
- Spiliotaki, M.; Mavroudis, D.; Kapranou, K.; Markomanolaki, H.; Kallergi, G.; Koinis, F.; Kalbakis, K.; Georgoulias, V.; Agelaki, S. Evaluation of proliferation and apoptosis markers in circulating tumor cells of women with early breast cancer who are candidates for tumor dormancy. Breast Cancer Res. 2014, 16, 485. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.L.; Adams, D.K.; Stefansson, S.; Haudenschild, C.; Martin, S.S.; Charpentier, M.; Chumsri, S.; Cristofanilli, M.; Tang, C.-M.; Alpaugh, R.K. Mitosis in circulating tumor cells stratifies highly aggressive breast carcinomas. Breast Cancer Res. 2016, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, C.; Mu, Z.; Rossi, G.; Schiewer, M.J.; Nguyen, T.; Austin, L.; Capoluongo, E.; Knudsen, K.E.; Cristofanilli, M.; Fortina, P. Detection of Activating Estrogen Receptor Gene (ESR1) Mutations in Single Circulating Tumor Cells. Clin. Cancer Res. 2017, 23, 6086–6093. [Google Scholar] [CrossRef] [PubMed]
- Reijm, E.A.; Sieuwerts, A.M.; Smid, M.; Vries, J.B.-D.; Mostert, B.; Onstenk, W.; Peeters, D.; Dirix, L.Y.; Seynaeve, C.M.; Jager, A.; et al. An 8-gene mRNA expression profile in circulating tumor cells predicts response to aromatase inhibitors in metastatic breast cancer patients. BMC Cancer 2016, 16, 123. [Google Scholar] [CrossRef]
- Beije, N.; Sieuwerts, A.M.; Kraan, J.; Van, N.M.; Onstenk, W.; Vitale, S.R.; van der Vlugt-Daane, M.; Dirix, L.Y.; Brouwer, A.; Hamberg, P.; et al. Estrogen receptor mutations and splice variants determined in liquid biopsies from metastatic breast cancer patients. Mol. Oncol. 2018, 12, 48–57. [Google Scholar] [CrossRef] [PubMed]
- US Food & Drug Administration. Premarket Approval, P150044—Cobas EGFR MUTATION TEST V2. 2016. Available online: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P150044 (accessed on 4 January 2019).
- Bredemeier, M.; Edimiris, P.; Tewes, M.; Mach, P.; Aktas, B.; Schellbach, D.; Wagner, J.; Kimmig, R.; Kasimir-Bauer, S. Establishment of a multimarker qPCR panel for the molecular characterization of circulating tumor cells in blood samples of metastatic breast cancer patients during the course of palliative treatment. Oncotarget 2016, 7, 41677–41690. [Google Scholar] [CrossRef] [PubMed]
- Quantify Agreement with Kappa. Available online: https://graphpad.com/quickcalcs/kappa1/” (accessed on 18 December 2018).






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keup, C.; Storbeck, M.; Hauch, S.; Hahn, P.; Sprenger-Haussels, M.; Tewes, M.; Mach, P.; Hoffmann, O.; Kimmig, R.; Kasimir-Bauer, S. Cell-Free DNA Variant Sequencing Using CTC-Depleted Blood for Comprehensive Liquid Biopsy Testing in Metastatic Breast Cancer. Cancers 2019, 11, 238. https://doi.org/10.3390/cancers11020238
Keup C, Storbeck M, Hauch S, Hahn P, Sprenger-Haussels M, Tewes M, Mach P, Hoffmann O, Kimmig R, Kasimir-Bauer S. Cell-Free DNA Variant Sequencing Using CTC-Depleted Blood for Comprehensive Liquid Biopsy Testing in Metastatic Breast Cancer. Cancers. 2019; 11(2):238. https://doi.org/10.3390/cancers11020238
Chicago/Turabian StyleKeup, Corinna, Markus Storbeck, Siegfried Hauch, Peter Hahn, Markus Sprenger-Haussels, Mitra Tewes, Pawel Mach, Oliver Hoffmann, Rainer Kimmig, and Sabine Kasimir-Bauer. 2019. "Cell-Free DNA Variant Sequencing Using CTC-Depleted Blood for Comprehensive Liquid Biopsy Testing in Metastatic Breast Cancer" Cancers 11, no. 2: 238. https://doi.org/10.3390/cancers11020238
APA StyleKeup, C., Storbeck, M., Hauch, S., Hahn, P., Sprenger-Haussels, M., Tewes, M., Mach, P., Hoffmann, O., Kimmig, R., & Kasimir-Bauer, S. (2019). Cell-Free DNA Variant Sequencing Using CTC-Depleted Blood for Comprehensive Liquid Biopsy Testing in Metastatic Breast Cancer. Cancers, 11(2), 238. https://doi.org/10.3390/cancers11020238