Integrated Approaches for the Use of Large Datasets to Identify Rational Therapies for the Treatment of Lung Cancers

Abstract
:1. Introduction
2. Datasets
3. Approaches
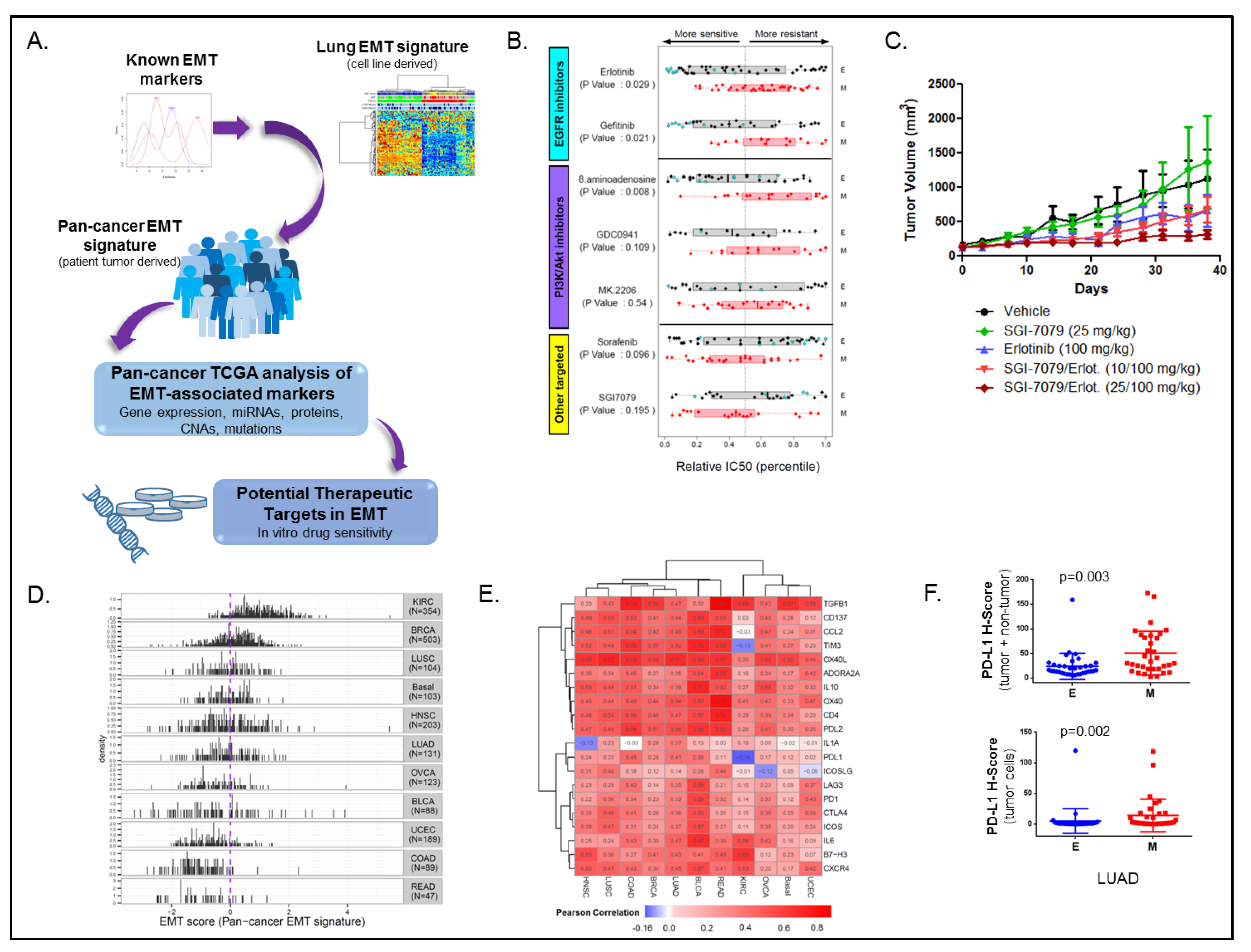
3.1. Cancer EMT Signature
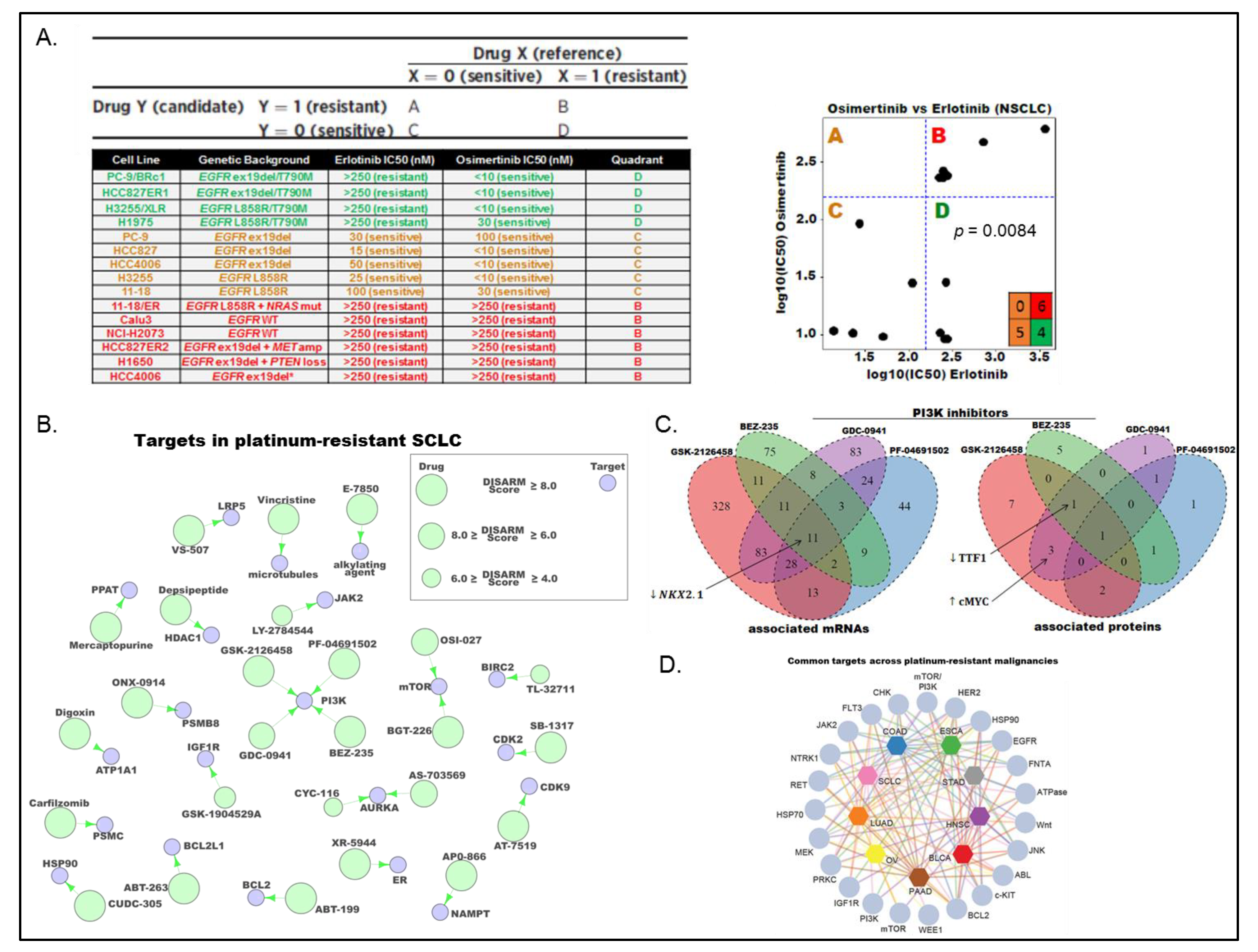
3.2. Proteomic Subgrouping of SCLC
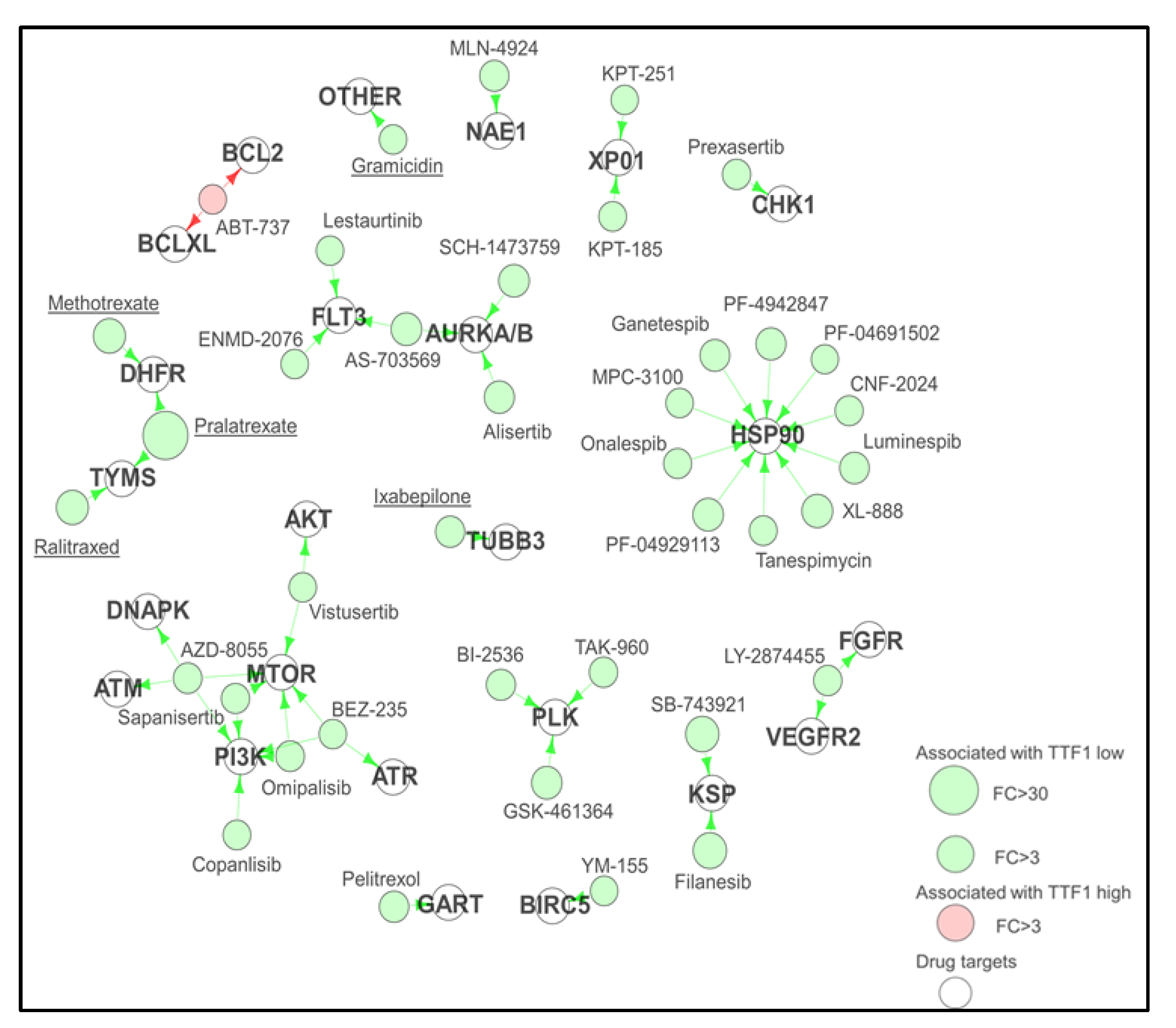
3.3. DISARM
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Tong, P.; Diao, L.; Shen, L.; Li, L.; Heymach, J.V.; Girard, L.; Minna, J.D.; Coombes, K.R.; Byers, L.A.; Wang, J. Selecting Reliable mRNA Expression Measurements Across Platforms Improves Downstream Analysis. Cancer Inform. 2016, 15, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Tong, P.; Coombes, K.R.; Johnson, F.M.; Byers, L.A.; Diao, L.; Liu, D.D.; Lee, J.J.; Heymach, J.V.; Wang, J. drexplorer: A tool to explore dose-response relationships and drug-drug interactions. Bioinformatics 2015, 31, 1692–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- University of Alabama School of Medicine. MacSynergy II. Available online: http://www.uab.edu/medicine/peds/macsynergy (accessed on 11 January 2019).
- Bliss, C. The toxicity of poisons applied jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Prichard, M.N.; Shipman, C., Jr. A three-dimensional model to analyze drug-drug interactions. Antivir. Res. 1990, 14, 181–205. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Kaneda, A.; Tsuji, S.; Isagawa, T.; Yamamoto, S.; Fujita, T.; Yamanaka, R.; Tanaka, Y.; Nukiwa, T.; Marquez, V.E.; et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci. Rep. 2013, 3, 1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TCGA. Available online: https://cancergenome.nih.gov/ (accessed on 11 January 2019).
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 11 January 2019).
- CCLE. Available online: https://portals.broadinstitute.org/ccle (accessed on 11 January 2019).
- GDSC. Available online: https://www.cancerrxgene.org/ (accessed on 11 January 2019).
- NCI-DTP. Available online: http://sclccelllines.cancer.gov/ (accessed on 11 January 2019).
- DISARM. Available online: http://ibl.mdanderson.org/DISARM/index.html (accessed on 11 January 2019).
- Larue, L.; Bellacosa, A. Epithelial–mesenchymal transition in development and cancer: Role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005, 24, 7443. [Google Scholar] [CrossRef]
- Radisky, D.C. Epithelial-mesenchymal transition. J. Cell Sci. 2005, 118, 4325–4326. [Google Scholar] [CrossRef] [Green Version]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef]
- Kim, E.S.; Herbst, R.S.; Wistuba, I.I.; Lee, J.J.; Blumenschein, G.R., Jr.; Tsao, A.; Stewart, D.J.; Hicks, M.E.; Erasmus, J., Jr.; Gupta, S.; et al. The BATTLE trial: Personalizing therapy for lung cancer. Cancer Discov. 2011, 1, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Kadara, H.; Behrens, C.; Liu, D.D.; Xiao, Y.; Rice, D.; Gazdar, A.F.; Fujimoto, J.; Moran, C.; Varella-Garcia, M.; et al. Abnormalities of the TITF-1 lineage-specific oncogene in NSCLC: Implications in lung cancer pathogenesis and prognosis. Clin. Cancer Res. 2011, 17, 2434–2443. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.; Hofman, V.; Dietel, M.; Soria, J.C.; Hofman, P. Assessment of the PD-L1 status by immunohistochemistry: Challenges and perspectives for therapeutic strategies in lung cancer patients. Virchows Arch. 2016, 468, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Tabchy, A.; Hennessy, B.T.; Gonzalez-Angulo, A.M.; Bernstam, F.M.; Lu, Y.; Mills, G.B. Quantitative proteomic analysis in breast cancer. Drugs Today 2011, 47, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Tibes, R.; Qiu, Y.; Lu, Y.; Hennessy, B.; Andreeff, M.; Mills, G.B.; Kornblau, S.M. Reverse phase protein array: Validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther. 2006, 5, 2512–2521. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ling, S.; Hegde, A.M.; Byers, L.A.; Coombes, K.; Mills, G.B.; Akbani, R. Using reverse-phase protein arrays as pharmacodynamic assays for functional proteomics, biomarker discovery, and drug development in cancer. Semin. Oncol. 2016, 43, 476–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagouri, F.; Bago-Horvath, Z.; Rossler, F.; Brandstetter, A.; Bartsch, R.; Papadimitriou, C.A.; Dimitrakakis, C.; Tsigginou, A.; Papaspyrou, I.; Giannos, A.; et al. High MET expression is an adverse prognostic factor in patients with triple-negative breast cancer. Br. J. Cancer 2013, 108, 1100–1105. [Google Scholar] [CrossRef] [Green Version]
- Cardnell, R.J.; Feng, Y.; Mukherjee, S.; Diao, L.; Tong, P.; Stewart, C.A.; Masrorpour, F.; Fan, Y.; Nilsson, M.; Shen, Y.; et al. Activation of the PI3K/mTOR Pathway following PARP Inhibition in Small Cell Lung Cancer. PLoS ONE 2016, 11, e0152584. [Google Scholar] [CrossRef]
- Fraley, C.; Raferty, A. MCLUST: Software for model-based cluster analysis. J. Classif. 1999, 16, 297–306. [Google Scholar] [CrossRef]
- Fraley, C.; Raferty, A. Enhanced model-based clustering, density estimation, and discriminant analysis software: MCLUST. J. Classif. 2003, 20, 263–286. [Google Scholar] [CrossRef]
- Polley, E.; Kunkel, M.; Evans, D.; Silvers, T.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Selby, M.; Connelly, J.; et al. Small Cell Lung Cancer Screen of Oncology Drugs, Investigational Agents, and Gene and microRNA Expression. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardnell, R.J.; Li, L.; Sen, T.; Bara, R.; Tong, P.; Fujimoto, J.; Ireland, A.S.; Guthrie, M.R.; Bheddah, S.; Banerjee, U.; et al. Protein expression of TTF1 and cMYC define distinct molecular subgroups of small cell lung cancer with unique vulnerabilities to aurora kinase inhibition, DLL3 targeting, and other targeted therapies. Oncotarget 2017, 8, 73419–73432. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955-961. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Manfredi, M.; Ecsedy, J.A. Scientific Rationale Supporting the Clinical Development Strategy for the Investigational Aurora A Kinase Inhibitor Alisertib in Cancer. Front. Oncol. 2015, 5, 189. [Google Scholar] [CrossRef]
- Melichar, B.; Adenis, A.; Lockhart, A.C.; Bennouna, J.; Dees, E.C.; Kayaleh, O.; Obermannova, R.; DeMichele, A.; Zatloukal, P.; Zhang, B.; et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: A five-arm phase 2 study. Lancet Oncol. 2015, 16, 395–405. [Google Scholar] [CrossRef]
- Owonikoko, T.; Nackaerts, K.; Scoszi, T.; Ostoros, G.; Baik, C.; Mark, Z.; Ullmann, C.D.; Niu, H.; Sheldon-Waniga, E.; Leonard, E.J.; et al. Randomized Phase 2 Study of the Investigational Aurora A Kinase (AAK) Inhibitor Alisertib (MLN8237) + Paclitaxel versus Placebo + Paclitaxel as Second-Line Therapy for Small Cell Lung Cancer (SCLC). Ann. Oncol. 2016, 27. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Holbeck, S.L.; Collins, J.M.; Doroshow, J.H. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol. Cancer Ther. 2010, 9, 1451–1460. [Google Scholar] [CrossRef]
- Teicher, B.A.; Silvers, T.; Selby, M.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Parchment, R.; Krushkal, J.; Sonkin, D.; et al. Small cell lung carcinoma cell line screen of etoposide/carboplatin plus a third agent. Cancer Med. 2017, 6, 1952–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, C.M.; Tong, P.; Cardnell, R.J.; Sen, T.; Su, X.; Ma, J.; Bara, R.O.; Johnson, F.M.; Wakefield, C.; Heymach, J.V.; et al. Differential Sensitivity Analysis for Resistant Malignancies (DISARM) Identifies Common Candidate Therapies across Platinum-Resistant Cancers. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- NCCN. Available online: http://www.nccn.org/professionals/physician_gls/pdf/sclc.pdf (accessed on 11 January 2019).
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic Profiling Identifies Dysregulated Pathways in Small Cell Lung Cancer and Novel Therapeutic Targets Including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCCN. Available online: https://www.nccn.org/professionals/physician_gls/default.aspx (accessed on 11 January 2019).
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Papadimitrakopoulou, V.; Lee, J.J.; Wistuba, I.I.; Tsao, A.S.; Fossella, F.V.; Kalhor, N.; Gupta, S.; Byers, L.A.; Izzo, J.G.; Gettinger, S.N.; et al. The BATTLE-2 Study: A Biomarker-Integrated Targeted Therapy Study in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3638–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardnell, R.J.; Feng, Y.; Diao, L.; Fan, Y.H.; Masrorpour, F.; Wang, J.; Shen, Y.; Mills, G.B.; Minna, J.D.; Heymach, J.V.; et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin. Cancer Res. 2013, 19, 6322–6328. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.A.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y.; et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017, 8, 28575–28587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietanza, M.C.; Waqar, S.N.; Krug, L.M.; Dowlati, A.; Hann, C.L.; Chiappori, A.; Owonikoko, T.K.; Woo, K.M.; Cardnell, R.J.; Fujimoto, J.; et al. Randomized, Double-Blind, Phase II Study of Temozolomide in Combination with Either Veliparib or Placebo in Patients with Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2386–2394. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resource | Malignancy | Data Types | Pre-Clinical/Clinical | Approach |
|---|---|---|---|---|
| TCGA [9] | Various | Genomic, transcriptomic, methylation, copy number, proteomic, and clinical # | Clinical | EMT |
| NCBI * [10] | Various | Genomic, transcriptomic, methylation, copy number and clinical # | Both | EMT, SCLC subgroups |
| CCLE [11] | Various | Drug sensitivity, genomic, and transcriptomic | Pre-clinical | SCLC subgroups, DISARM |
| GDSC [12] | Various | Drug sensitivity, genomic, and transcriptomic | Pre-clinical | SCLC subgroups, DISARM |
| NCI Developmental Therapeutics Program [13] | SCLC | Drug sensitivity, and transcriptomic | Pre-clinical | SCLC subgroups, DISARM |
| DISARM [14] | Various | Drug sensitivity | Pre-clinical | DISARM |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardnell, R.J.; Byers, L.A.; Wang, J. Integrated Approaches for the Use of Large Datasets to Identify Rational Therapies for the Treatment of Lung Cancers. Cancers 2019, 11, 239. https://doi.org/10.3390/cancers11020239
Cardnell RJ, Byers LA, Wang J. Integrated Approaches for the Use of Large Datasets to Identify Rational Therapies for the Treatment of Lung Cancers. Cancers. 2019; 11(2):239. https://doi.org/10.3390/cancers11020239
Chicago/Turabian StyleCardnell, Robert J., Lauren Averett Byers, and Jing Wang. 2019. "Integrated Approaches for the Use of Large Datasets to Identify Rational Therapies for the Treatment of Lung Cancers" Cancers 11, no. 2: 239. https://doi.org/10.3390/cancers11020239
APA StyleCardnell, R. J., Byers, L. A., & Wang, J. (2019). Integrated Approaches for the Use of Large Datasets to Identify Rational Therapies for the Treatment of Lung Cancers. Cancers, 11(2), 239. https://doi.org/10.3390/cancers11020239