Facets of Communication: Gap Junction Ultrastructure and Function in Cancer Stem Cells and Tumor Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
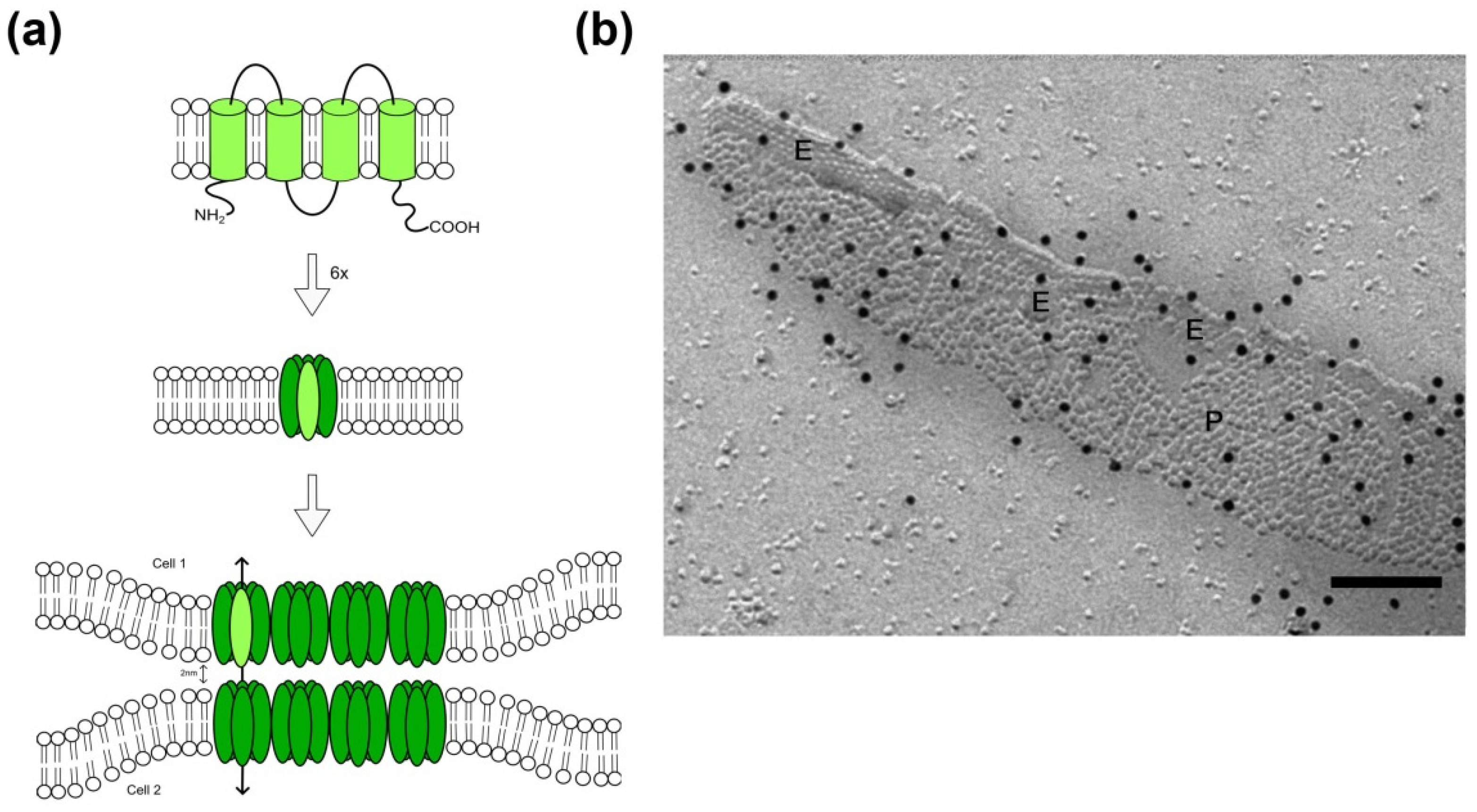
2. Gap Junction Proteins
3. Gap junction Morphology in Carcinogenesis
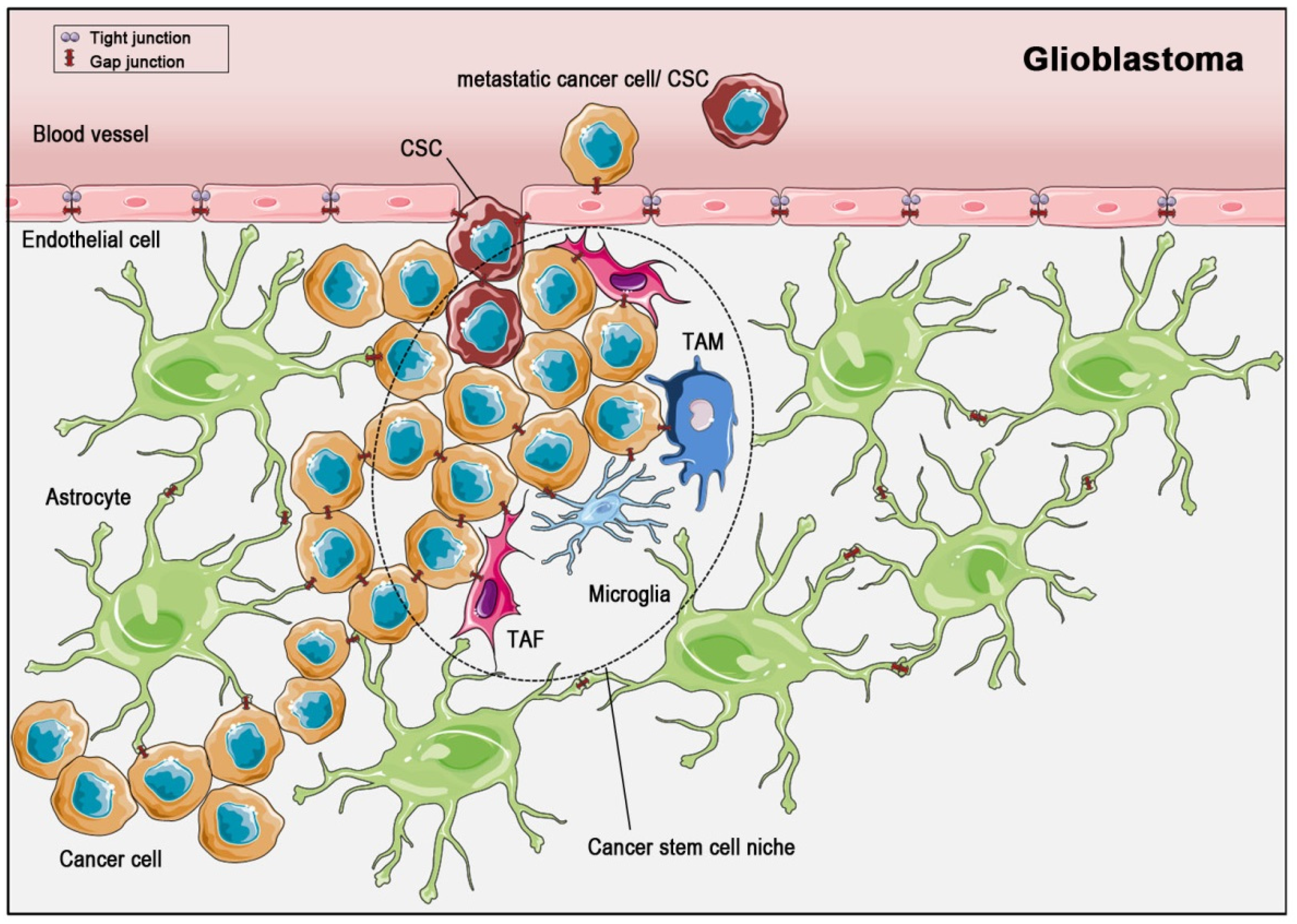
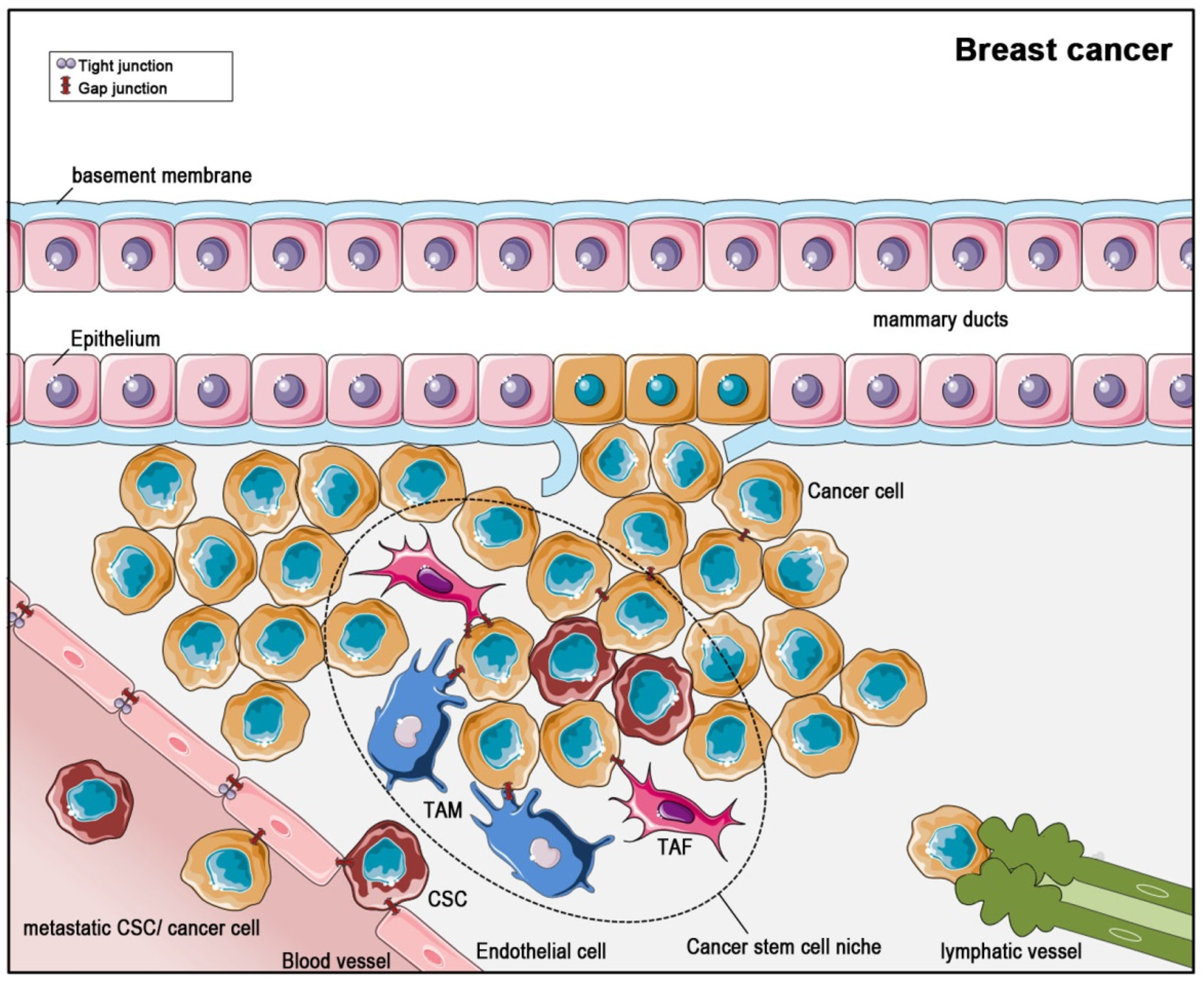
4. Gap Junctions in Solid Tumors—Glioblastoma and Breast Cancer Visited Exemplarily
5. Cancer Stem Cells
6. Connexins in Cancer Stem Cells
7. Cancer Stem Cells, the Stem Cell Niche and Therapeutic Approaches
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Cancer—Key Facts. Available online: http://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 18 September 2018).
- Garber, J.E.; Offit, K. Hereditary cancer predisposition syndromes. J. Clin. Oncol. 2005, 23, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T. Formation of UV-induced DNA damage contributing to skin cancer development. Photochem. Photobiol. Sci. 2018, 17, 1816–1841. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Thaiparambil, J.; Donepudi, S.R.; Vantaku, V.; Piyarathna, D.W.B.; Maity, S.; Krishnapuram, R.; Putluri, V.; Gu, F.; Purwaha, P.; et al. Tobacco-Specific Carcinogens Induce Hypermethylation, DNA Adducts, and DNA Damage in Bladder Cancer. Cancer Prev. Res. 2017, 10, 588–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, S.; Vu, K.; Tai, P.; Joseph, K.; Koul, R.; Dubey, A.; Yu, E. Radiation-induced second malignancies. Anticancer Res. 2015, 35, 2431–2434. [Google Scholar] [PubMed]
- Varmus, H.; Pao, W.; Politi, K.; Podsypanina, K.; Du, Y.C.N. Oncogenes come of age. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kashuba, V.I.; Pavlova, T.V.; Grigorieva, E.V.; Kutsenko, A.; Yenamandra, S.P.; Li, J.; Wang, F.; Protopopov, A.I.; Zabarovska, V.I.; Senchenko, V.; et al. High mutability of the tumor suppressor genes RASSF1 and RBSP3 (CTDSPL) in cancer. PLoS ONE 2009, 4, e5231. [Google Scholar] [CrossRef] [PubMed]
- de Miranda, N.F.C.C.; Peng, R.; Georgiou, K.; Wu, C.; Falk Sörqvist, E.; Berglund, M.; Chen, L.; Gao, Z.; Lagerstedt, K.; Lisboa, S.; et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J. Exp. Med. 2013, 210, 1729–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slattery, M.L.; Herrick, J.S.; Mullany, L.E.; Samowitz, W.S.; Sevens, J.R.; Sakoda, L.; Wolff, R.K. The co-regulatory networks of tumor suppressor genes, oncogenes, and miRNAs in colorectal cancer. Genes Chromosom. Cancer 2017, 56, 769–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.X.; Massagué, J. Genetic determinants of cancer metastasis. Nat. Rev. Genet. 2007, 8, 341. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E.; Ruch, R.J. Cell-cell communication in carcinogenesis. Front. Biosci. 1998, 3, d208–d236. [Google Scholar] [CrossRef] [PubMed]
- Mesnil, M.; Crespin, S.; Avanzo, J.-L.; Zaidan-Dagli, M.-L. Defective gap junctional intercellular communication in the carcinogenic process. Biochim. Biophys. Acta 2005, 1719, 125–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawaz, M.; Fatima, F. Extracellular Vesicles, Tunneling Nanotubes, and Cellular Interplay: Synergies and Missing Links. Front. Mol. Biosci. 2017, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.H.; Martin, P.E. Gap junctions: Structure and function (Review). Mol. Membr. Biol. 2002, 19, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and the control of tissue growth: Lack of communication between cancer cells. Nature 1966, 209, 1248–1249. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and tissue growth. I. Cancerous growth. J. Cell Biol. 1967, 33, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Cronier, L.; Crespin, S.; Strale, P.O.; Defamie, N.; Mesnil, M. Gap junctions and cancer: New functions for an old story. Antioxid. Redox Signal. 2009, 11, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Holder, J.W.; Elmore, E.; Barrett, J.C. Gap junction function and cancer. Cancer Res. 1993, 53, 3475–3485. [Google Scholar] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Benko, G.; Spajic, B.; Demirovic, A.; Stimac, G.; Kru Sbreve Lin, B.; Tomas, D. Prognostic value of connexin43 expression in patients with clinically localized prostate cancer. Prostate Cancer Prostatic Dis. 2011, 14, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.L.; Wang, B.R.; Chen, G.Q.; Li, G.H.; Xu, Y.Y. Clinical significance of vascular endothelial growth factor and connexin43 for predicting pancreatic cancer clinicopathologic parameters. Med. Oncol. 2010, 27, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Teleki, I.; Szasz, A.M.; Maros, M.E.; Gyorffy, B.; Kulka, J.; Meggyeshazi, N.; Kiszner, G.; Balla, P.; Samu, A.; Krenacs, T. Correlations of differentially expressed gap junction connexins Cx26, Cx30, Cx32, Cx43 and Cx46 with breast cancer progression and prognosis. PLoS ONE 2014, 9, e112541. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Maeda, K.; Noda, E.; Inoue, T.; Fukunaga, S.; Nagahara, H.; Hirakawa, K. Clinical significance of the expression of connexin26 in colorectal cancer. J. Exp. Clin. Cancer Res. 2010, 29, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Furuya, T.; Li, D.; Xu, J.; Cao, X.; Li, Q.; Xu, J.; Xu, Z.; Sasaki, K.; Liu, X. Connexin 26 expression correlates with less aggressive phenotype of intestinal type-gastric carcinomas. Int. J. Mol. Med. 2010, 25, 709–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyet, C.; Buser, L.; Roudnicky, F.; Detmar, M.; Hermanns, T.; Mannhard, D.; Hohn, A.; Ruschoff, J.; Zhong, Q.; Sulser, T.; et al. Connexin 43 expression predicts poor progression-free survival in patients with non-muscle invasive urothelial bladder cancer. J. Clin. Pathol. 2015, 68, 819–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teleki, I.; Krenacs, T.; Szasz, M.A.; Kulka, J.; Wichmann, B.; Leo, C.; Papassotiropoulos, B.; Riemenschnitter, C.; Moch, H.; Varga, Z. The potential prognostic value of connexin 26 and 46 expression in neoadjuvant-treated breast cancer. BMC Cancer 2013, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Ezumi, K.; Yamamoto, H.; Murata, K.; Higashiyama, M.; Damdinsuren, B.; Nakamura, Y.; Kyo, N.; Okami, J.; Ngan, C.Y.; Takemasa, I.; et al. Aberrant expression of connexin 26 is associated with lung metastasis of colorectal cancer. Clin. Cancer Res. 2008, 14, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Naoi, Y.; Miyoshi, Y.; Taguchi, T.; Kim, S.J.; Arai, T.; Maruyama, N.; Tamaki, Y.; Noguchi, S. Connexin26 expression is associated with aggressive phenotype in human papillary and follicular thyroid cancers. Cancer Lett. 2008, 262, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim. Biophys. Acta 2005, 1711, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solan, J.L.; Lampe, P.D. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim. Biophys. Acta 2005, 1711, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.I.; Wang, L.H. Emerging roles of gap junction proteins connexins in cancer metastasis, chemoresistance and clinical application. J. Biomed. Sci. 2019, 26, 8. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V.; Jiang, J.X.; Mesnil, M. Connexins and Pannexins: Important Players in Tumorigenesis, Metastasis and Potential Therapeutics. Int. J. Mol. Sci. 2018, 19, 1645. [Google Scholar] [CrossRef] [PubMed]
- Sinyuk, M.; Mulkearns-Hubert, E.E.; Reizes, O.; Lathia, J. Cancer Connectors: Connexins, Gap Junctions, and Communication. Front. Oncol. 2018, 8, 646. [Google Scholar] [CrossRef] [PubMed]
- Borek, C.; Higashino, S.; Loewenstein, W.R. Intercellular communication and tissue growth: IV. Conductance of membrane junctions of normal and cancerous cells in culture. J. Membr. Biol. 1969, 1, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Sohl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saez, J.C.; Retamal, M.A.; Basilio, D.; Bukauskas, F.F.; Bennett, M.V. Connexin-based gap junction hemichannels: Gating mechanisms. Biochim. Biophys. Acta 2005, 1711, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [PubMed]
- Revel, J.P.; Karnovsky, M.J. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J. Cell Biol. 1967, 33, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Peracchia, C. Gap junctions. Structural changes after uncoupling procedures. J. Cell Biol. 1977, 72, 628–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, K. Freeze-fracture replica electron microscopy combined with SDS digestion for cytochemical labeling of integral membrane proteins. Application to the immunogold labeling of intercellular junctional complexes. J. Cell Sci. 1995, 108, 3443–3449. [Google Scholar] [PubMed]
- Rash, J.E.; Duffy, H.S.; Dudek, F.E.; Bilhartz, B.L.; Whalen, L.R.; Yasumura, T. Grid-mapped freeze-fracture analysis of gap junctions in gray and white matter of adult rat central nervous system, with evidence for a “panglial syncytium” that is not coupled to neurons. J. Comp. Neurol. 1997, 388, 265–292. [Google Scholar] [CrossRef]
- Meier, C.; Beckmann, A. Freeze fracture: New avenues for the ultrastructural analysis of cells in vitro. Histochem. Cell Biol. 2018, 149, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Panchin, Y.; Kelmanson, I.; Matz, M.; Lukyanov, K.; Usman, N.; Lukyanov, S. A ubiquitous family of putative gap junction molecules. Curr. Biol. 2000, 10, R473–R474. [Google Scholar] [CrossRef]
- Dahl, G.; Muller, K.J. Innexin and pannexin channels and their signaling. FEBS Lett. 2014, 588, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penuela, S.; Gehi, R.; Laird, D.W. The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 2013, 1828, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosi, C.; Gassmann, O.; Pranskevich, J.N.; Boassa, D.; Smock, A.; Wang, J.; Dahl, G.; Steinem, C.; Sosinsky, G.E. Pannexin1 and Pannexin2 channels show quaternary similarities to connexons and different oligomerization numbers from each other. J. Biol. Chem. 2010, 285, 24420–24431. [Google Scholar] [CrossRef] [PubMed]
- Boassa, D.; Ambrosi, C.; Qiu, F.; Dahl, G.; Gaietta, G.; Sosinsky, G. Pannexin1 channels contain a glycosylation site that targets the hexamer to the plasma membrane. J. Biol. Chem. 2007, 282, 31733–31743. [Google Scholar] [CrossRef] [PubMed]
- Sosinsky, G.E.; Boassa, D.; Dermietzel, R.; Duffy, H.S.; Laird, D.W.; MacVicar, B.; Naus, C.C.; Penuela, S.; Scemes, E.; Spray, D.C.; et al. Pannexin channels are not gap junction hemichannels. Channels 2011, 5, 193–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckmann, A.; Grissmer, A.; Krause, E.; Tschernig, T.; Meier, C. Pannexin-1 channels show distinct morphology and no gap junction characteristics in mammalian cells. Cell Tissue Res. 2016, 363, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.G.; Reynhout, J.K.; TenBroek, E.M.; Quade, B.J.; Yasumura, T.; Davidson, K.G.V.; Sheridan, J.D.; Rash, J.E. Gap junction assembly: Roles for the formation plaque and regulation by the C-terminus of connexin43. Mol. Biol. Cell 2012, 23, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, R.; Hormuzdi, S.G.; Barbe, M.T.; Herb, A.; Monyer, H. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13644–13649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, G.; Locovei, S. Pannexin: To gap or not to gap, is that a question? IUBMB Life 2006, 58, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef] [PubMed]
- Locovei, S.; Wang, J.; Dahl, G. Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett. 2006, 580, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Locovei, S.; Dahl, G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004, 572, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, G. The Pannexin1 membrane channel: Distinct conformations and functions. FEBS Lett. 2018, 592, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musil, L.S.; Le, A.C.; VanSlyke, J.K.; Roberts, L.M. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J. Biol. Chem. 2000, 275, 25207–25215. [Google Scholar] [CrossRef] [PubMed]
- Lauf, U.; Giepmans, B.N.; Lopez, P.; Braconnot, S.; Chen, S.C.; Falk, M.M. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 10446–10451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolburg, H.; Rohlmann, A. Structure-Function Relationships in Gap Junctions. In International Review of Cytology; Jeon, K.W., Jarvik, J., Eds.; Academic Press: Cambridge, MA, USA, 1995; Volume 157, pp. 315–373. [Google Scholar]
- Solan, J.L.; Lampe, P.D. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta 2018, 1860, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.W.; Barker, R.J.; Zhu, C.; Gourdie, R.G. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol. Biol. Cell 2005, 16, 5686–5698. [Google Scholar] [CrossRef] [PubMed]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.; Chodock, R.; Hand, A.R.; Laird, D.W. The origin of annular junctions: A mechanism of gap junction internalization. J. Cell Sci. 2001, 114, 763–773. [Google Scholar] [PubMed]
- Norris, R.P.; Baena, V.; Terasaki, M. Localization of phosphorylated connexin 43 using serial section immunogold electron microscopy. J. Cell Sci. 2017, 130, 1333. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.A.; Lampe, P.D. Injury-triggered Akt phosphorylation of Cx43: A ZO-1-driven molecular switch that regulates gap junction size. J. Cell Sci. 2014, 127, 455–464. [Google Scholar] [CrossRef] [PubMed]
- McNutt, N.S.; Weinstein, R.S. Carcinoma of the cervix: Deficiency of nexus intercellular junctions. Science 1969, 165, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Priollet, B.C.; Raison, D.; Molinie, V.; Guillausseau, P.J.; Wassef, M.; Bouchaud, C. Altered Gap and Tight Junctions in Human Thyroid Oncocytic Tumors: A Study of 8 Cases by Freeze-fracture. Ultrastruct. Pathol. 1998, 22, 413–420. [Google Scholar] [CrossRef]
- Tada, J.; Hashimoto, K. Ultrastructural localization of gap junction protein connexin 43 in normal human skin, basal cell carcinoma, and squamous cell carcinoma. J. Cutan. Pathol. 1997, 24, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Schenk, P. Gap junctions in laryngeal carcinoma. Arch. Oto-Rhino-Laryngol. 1980, 228, 51–56. [Google Scholar] [CrossRef]
- Kirichenko, E.Y.; Savchenko, A.F.; Kozachenko, D.V.; Matsionis, A.E.; Logvinov, A.K. Ultrastructural characteristics of gap junctions in human glial brain tumors. Arkhiv Patologii 2017, 79, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Veruki, M.L.; Bukoreshtliev, N.V.; Hartveit, E.; Gerdes, H.-H. Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc. Natl. Acad. Sci. USA 2010, 107, 17194–17199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tani, E.; Ikeda, K.; Yamagata, S.; Nishiura, M.; Higashi, N. Specialized junctional complexes in human meningioma. Acta Neuropathol. 1974, 28, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Arishima, H.; Sato, K.; Kubota, T. Immunohistochemical and Ultrastructural Study of Gap Junction Proteins Connexin26 and 43 in Human Arachnoid Villi and Meningeal Tumors. J. Neuropathol. Exp. Neurol. 2002, 61, 1048–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovcevska, I.; Kocevar, N.; Komel, R. Glioma and glioblastoma - how much do we (not) know? Mol. Clin. Oncol. 2013, 1, 935–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee Sh, U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Balca-Silva, J.; Matias, D.; Dubois, L.G.; Carneiro, B.; do Carmo, A.; Girao, H.; Ferreira, F.; Ferrer, V.P.; Chimelli, L.; Filho, P.N.; et al. The Expression of Connexins and SOX2 Reflects the Plasticity of Glioma Stem-Like Cells. Transl. Oncol. 2017, 10, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Chasampalioti, M.; Green, A.R.; Ellis, I.O.; Rakha, E.A.; Jackson, A.M.; Spendlove, I.; Ramage, J.M. Connexin 43 is an independent predictor of patient outcome in breast cancer patients. Breast Cancer Res. Treat. 2018. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Rodriguez-Cruz, V.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis. 2014, 5, e1145. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.F.; Varghese, R.T.; Lamouille, S.; Guo, S.; Pridham, K.J.; Kanabur, P.; Osimani, A.M.; Sharma, S.; Jourdan, J.; Rodgers, C.M.; et al. Connexin 43 Inhibition Sensitizes Chemoresistant Glioblastoma Cells to Temozolomide. Cancer Res. 2016, 76, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, M.; Deleyrolle, L.P.; Mulkearns-Hubert, E.E.; Jarrar, A.; Li, M.; Sinyuk, M.; Otvos, B.; Brunet, S.; Flavahan, W.A.; Hubert, C.G.; et al. Differential connexin function enhances self-renewal in glioblastoma. Cell Rep. 2015, 11, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, R.; Barbe, M.T.; Jakob, N.J.; Monyer, H. Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J. Neurochem. 2005, 92, 1033–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, J.S.; Baumgarten, I.M.; Harley, E.H. Reversible inhibition of intercellular junctional communication by glycyrrhetinic acid. Biochem. Biophys. Res. Commun. 1986, 134, 29–36. [Google Scholar] [CrossRef]
- Owrang, M.; Copeland, R.L., Jr.; Ricks-Santi, L.J.; Gaskins, M.; Beyene, D.; Dewitty, R.L., Jr.; Kanaan, Y.M. Breast Cancer Prognosis for Young Patients. In Vivo 2017, 31, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.L.; Williams, C.B.; Zambrano, J.N.; Williams, C.J.; Yeh, E.S. Connexin 43 in the development and progression of breast cancer: What’s the connection? (Review). Int. J. Oncol. 2017, 51, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Kanczuga-Koda, L.; Sulkowski, S.; Lenczewski, A.; Koda, M.; Wincewicz, A.; Baltaziak, M.; Sulkowska, M. Increased expression of connexins 26 and 43 in lymph node metastases of breast cancer. J. Clin. Pathol. 2006, 59, 429–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.X.; Penuela, S. Connexin and pannexin channels in cancer. BMC Cell Biol. 2016, 17 (Suppl. 1), 12. [Google Scholar] [CrossRef] [PubMed]
- Cowan, K.N.; Langlois, S.; Penuela, S.; Cowan, B.J.; Laird, D.W. Pannexin1 and Pannexin3 exhibit distinct localization patterns in human skin appendages and are regulated during keratinocyte differentiation and carcinogenesis. Cell Commun. Adhes. 2012, 19, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.P.; Bechberger, J.F.; Thompson, R.J.; MacVicar, B.A.; Bruzzone, R.; Naus, C.C. Tumor-suppressive effects of pannexin 1 in C6 glioma cells. Cancer Res. 2007, 67, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.K.; Plante, I.; Penuela, S.; Laird, D.W. Loss of Panx1 Impairs Mammary Gland Development at Lactation: Implications for Breast Tumorigenesis. PLoS ONE 2016, 11, e0154162. [Google Scholar] [CrossRef] [PubMed]
- Ebert, R.; Meissner-Weigl, J.; Zeck, S.; Maatta, J.; Auriola, S.; Coimbra de Sousa, S.; Mentrup, B.; Graser, S.; Rachner, T.D.; Hofbauer, L.C.; et al. Probenecid as a sensitizer of bisphosphonate-mediated effects in breast cancer cells. Mol. Cancer 2014, 13, 265. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Guzman, M.E.; Bigoni-Ordonez, G.D.; Ibanez Hernandez, M.; Ortiz-Sanchez, E. Cancer stem cell impact on clinical oncology. World J. Stem Cells 2018, 10, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Medema, J.P. Cancer stem cells—Important players in tumor therapy resistance. FEBS J. 2014, 281, 4779–4791. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Farace, C.; Oliver, J.A.; Melguizo, C.; Alvarez, P.; Bandiera, P.; Rama, A.R.; Malaguarnera, G.; Ortiz, R.; Madeddu, R.; Prados, J. Microenvironmental Modulation of Decorin and Lumican in Temozolomide-Resistant Glioblastoma and Neuroblastoma Cancer Stem-Like Cells. PLoS ONE 2015, 10, e0134111. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology 2014, 28, 1101–1107. [Google Scholar] [PubMed]
- Schulenburg, A.; Blatt, K.; Cerny-Reiterer, S.; Sadovnik, I.; Herrmann, H.; Marian, B.; Grunt, T.W.; Zielinski, C.C.; Valent, P. Cancer stem cells in basic science and in translational oncology: Can we translate into clinical application? J. Hematol. Oncol. 2015, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskarsson, T.; Batlle, E.; Massagué, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Wu, D.; Wu, P.; Chen, Z.; Huang, J. The cancer stem cell niche: Cross talk between cancer stem cells and their microenvironment. Tumour Biol. 2014, 35, 3945–3951. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M. Extracellular vesicle-mediated transport of non-coding RNAs between stem cells and cancer cells: Implications in tumor progression and therapeutic resistance. Stem Cell Investig. 2017, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Moghbeli, M.; Moghbeli, F.; Forghanifard, M.M.; Abbaszadegan, M.R. Cancer stem cell detection and isolation. Med. Oncol. 2014, 31, 69. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E. Cancer Prevention and Therapy of Two Types of Gap Junctional Intercellular Communication(-)Deficient “Cancer Stem Cell”. Cancers 2019, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ginestier, C.; Ou, S.J.; Clouthier, S.G.; Patel, S.H.; Monville, F.; Korkaya, H.; Heath, A.; Dutcher, J.; Kleer, C.G.; et al. Breast Cancer Stem Cells Are Regulated by Mesenchymal Stem Cells through Cytokine Networks. Cancer Res. 2011, 71, 614–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Borovski, T.; De Sousa, E.M.F.; Vermeulen, L.; Medema, J.P. Cancer stem cell niche: The place to be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.Q.; Alexandrou, A.T.; Li, J.J. Breast cancer stem cells: Multiple capacities in tumor metastasis. Cancer Lett. 2014, 349, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiagarajan, P.S.; Sinyuk, M.; Turaga, S.M.; Mulkearns-Hubert, E.E.; Hale, J.S.; Rao, V.; Demelash, A.; Saygin, C.; China, A.; Alban, T.J.; et al. Cx26 drives self-renewal in triple-negative breast cancer via interaction with NANOG and focal adhesion kinase. Nat. Commun. 2018, 9, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samardzija, C.; Greening, D.W.; Escalona, R.; Chen, M.; Bilandzic, M.; Luwor, R.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. Knockdown of stem cell regulator Oct4A in ovarian cancer reveals cellular reprogramming associated with key regulators of cytoskeleton-extracellular matrix remodelling. Sci. Rep. 2017, 7, 46312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.C.; Xiao, H.L.; Jiang, X.F.; Wang, Q.L.; Li, Y.; Yang, X.J.; Ping, Y.F.; Duan, J.J.; Jiang, J.Y.; Ye, X.Z.; et al. Connexin 43 reverses malignant phenotypes of glioma stem cells by modulating E-cadherin. Stem Cells 2012, 30, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Gangoso, E.; Thirant, C.; Chneiweiss, H.; Medina, J.M.; Tabernero, A. A cell-penetrating peptide based on the interaction between c-Src and connexin43 reverses glioma stem cell phenotype. Cell Death Dis. 2014, 5, e1023. [Google Scholar] [CrossRef] [PubMed]
- Jaraiz-Rodriguez, M.; Tabernero, M.D.; Gonzalez-Tablas, M.; Otero, A.; Orfao, A.; Medina, J.M.; Tabernero, A. A Short Region of Connexin43 Reduces Human Glioma Stem Cell Migration, Invasion, and Survival through Src, PTEN, and FAK. Stem Cell Rep. 2017, 9, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Calalb, M.B.; Polte, T.R.; Hanks, S.K. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: A role for Src family kinases. Mol. Cell. Biol. 1995, 15, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Omori, Y.; Li, Q.; Nishikawa, Y.; Yoshioka, T.; Yoshida, M.; Ishikawa, K.; Enomoto, K. Cytoplasmic accumulation of connexin32 expands cancer stem cell population in human HuH7 hepatoma cells by enhancing its self-renewal. Int. J. Cancer 2011, 128, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Ruch, R.J. Connexin43 Suppresses Lung Cancer Stem Cells. Cancers 2019, 11, 175. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. AS602801, an Anti-Cancer Stem Cell Drug Candidate, Suppresses Gap-junction Communication Between Lung Cancer Stem Cells and Astrocytes. Anticancer Res. 2018, 38, 5093–5099. [Google Scholar] [CrossRef] [PubMed]
- Uzu, M.; Sin, W.C.; Shimizu, A.; Sato, H. Conflicting Roles of Connexin43 in Tumor Invasion and Growth in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 1159. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Mao, X.Y.; Li, Q.Q.; Gao, Y.F.; Zhou, H.H.; Liu, Z.Q.; Jin, W.L. Gap junction as an intercellular glue: Emerging roles in cancer EMT and metastasis. Cancer Lett. 2016, 381, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Elzarrad, M.K.; Haroon, A.; Willecke, K.; Dobrowolski, R.; Gillespie, M.N.; Al-Mehdi, A.B. Connexin-43 upregulation in micrometastases and tumor vasculature and its role in tumor cell attachment to pulmonary endothelium. BMC Med. 2008, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Stoletov, K.; Strnadel, J.; Zardouzian, E.; Momiyama, M.; Park, F.D.; Kelber, J.A.; Pizzo, D.P.; Hoffman, R.; VandenBerg, S.R.; Klemke, R.L. Role of connexins in metastatic breast cancer and melanoma brain colonization. J. Cell Sci. 2013, 126, 904–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabernero, A.; Gangoso, E.; Jaraiz-Rodriguez, M.; Medina, J.M. The role of connexin43-Src interaction in astrocytomas: A molecular puzzle. Neuroscience 2016, 323, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Z.; Jiang, J.X. Gap junction and hemichannel-independent actions of connexins on cell and tissue functions—An update. FEBS Lett. 2014, 588, 1186–1192. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beckmann, A.; Hainz, N.; Tschernig, T.; Meier, C. Facets of Communication: Gap Junction Ultrastructure and Function in Cancer Stem Cells and Tumor Cells. Cancers 2019, 11, 288. https://doi.org/10.3390/cancers11030288
Beckmann A, Hainz N, Tschernig T, Meier C. Facets of Communication: Gap Junction Ultrastructure and Function in Cancer Stem Cells and Tumor Cells. Cancers. 2019; 11(3):288. https://doi.org/10.3390/cancers11030288
Chicago/Turabian StyleBeckmann, Anja, Nadine Hainz, Thomas Tschernig, and Carola Meier. 2019. "Facets of Communication: Gap Junction Ultrastructure and Function in Cancer Stem Cells and Tumor Cells" Cancers 11, no. 3: 288. https://doi.org/10.3390/cancers11030288
APA StyleBeckmann, A., Hainz, N., Tschernig, T., & Meier, C. (2019). Facets of Communication: Gap Junction Ultrastructure and Function in Cancer Stem Cells and Tumor Cells. Cancers, 11(3), 288. https://doi.org/10.3390/cancers11030288