Mechanisms of Anticancer Drug Resistance in Hepatoblastoma

 ,
, 


{kind=link}
{kind=link}
Abstract
:1. Introduction
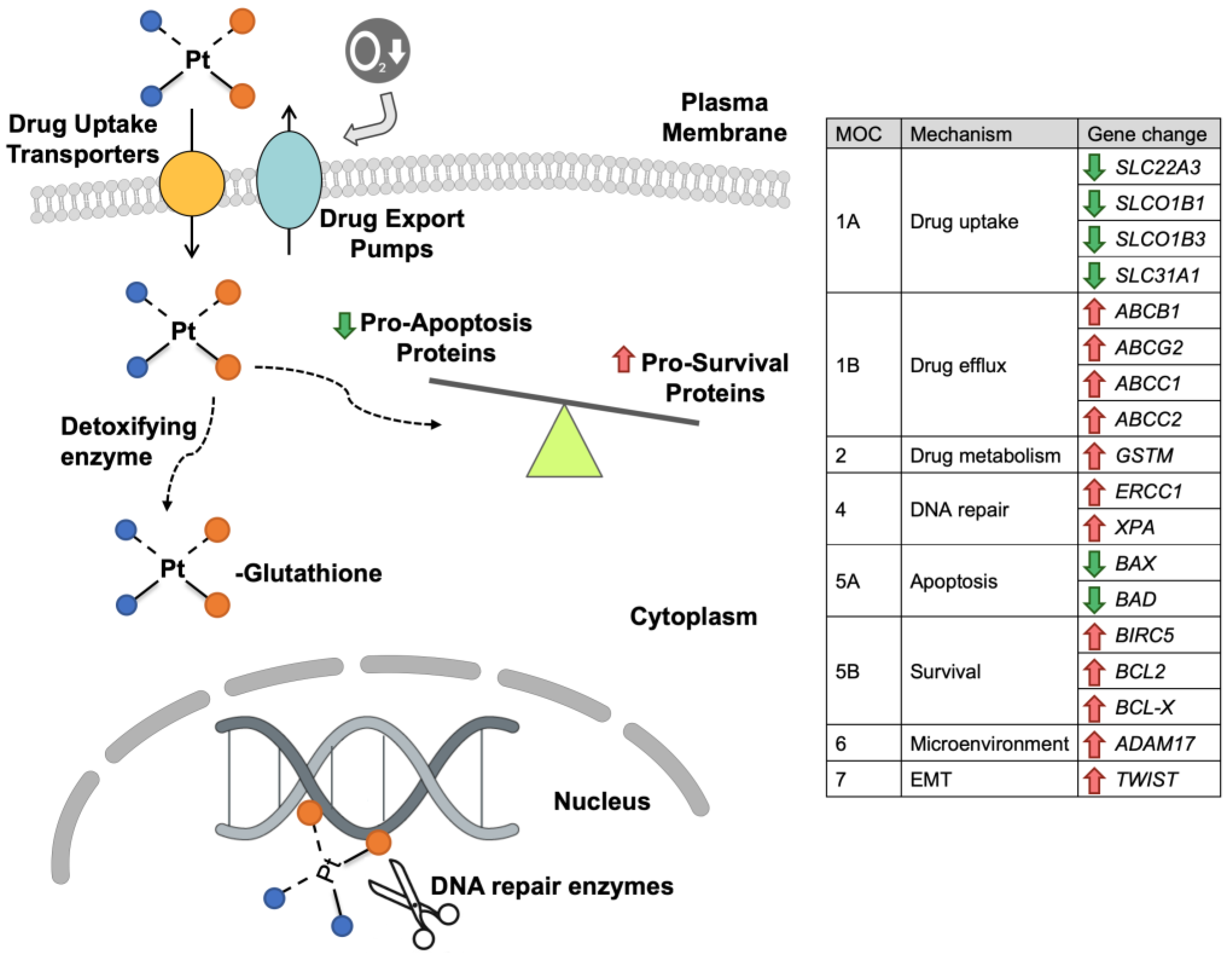
2. Mechanisms of HB Resistance to Platinum Derivatives
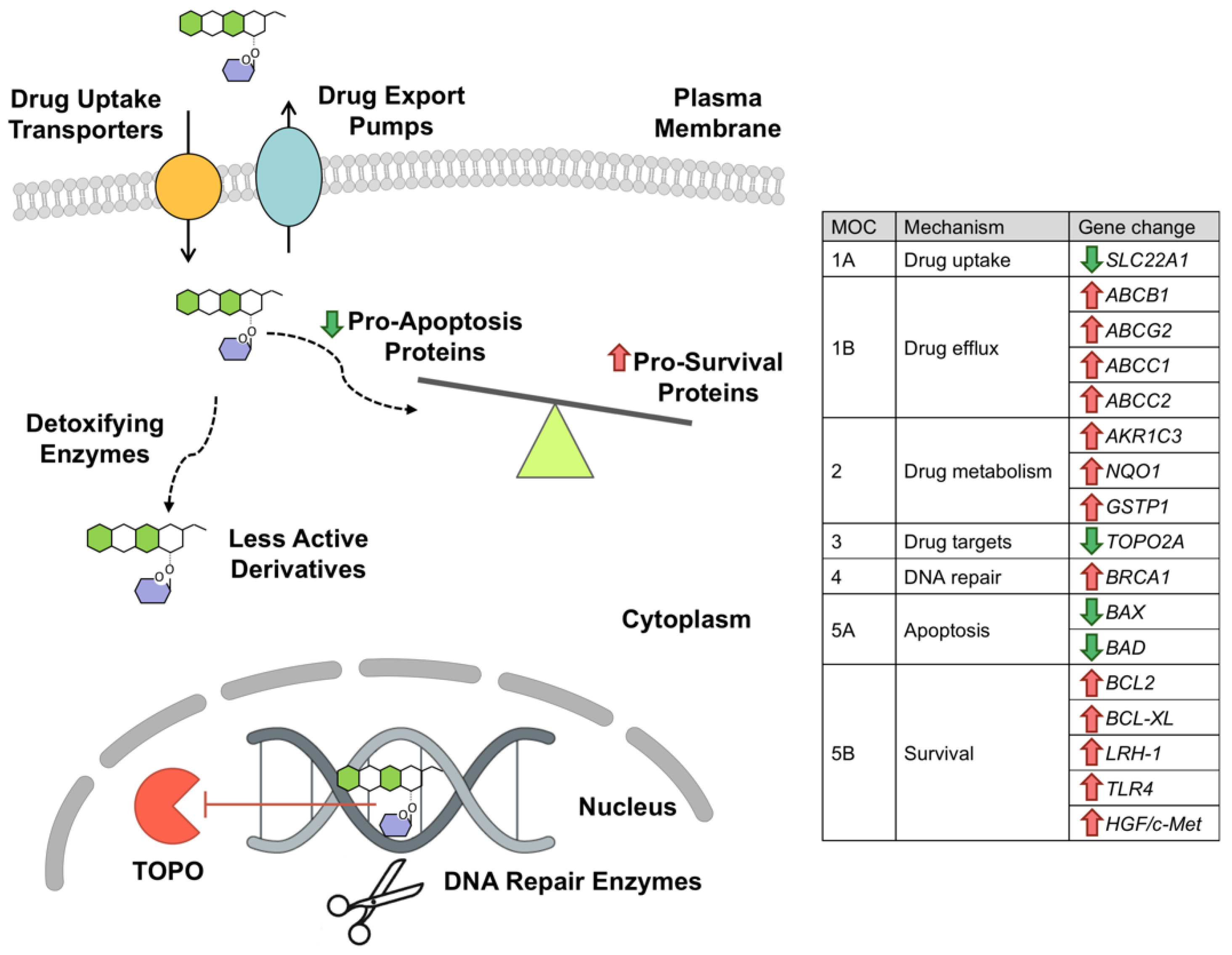
3. Mechanisms of HB Resistance to Anthracyclines
4. Mechanisms of HB Resistance to Etoposide
5. Mechanisms of HB Resistance to Tyrosine-Kinase Inhibitors (TKIs)
6. Mechanisms of HB Resistance to Vinca Alkaloids
7. Mechanisms of HB Resistance to 5-Fluorouracil (5-FU)
8. Mechanisms of HB Resistance to Nitrogen Mustards
9. Mechanisms of HB Resistance to Bevacizumab
10. Mechanisms of HB Resistance to Irinotecan
11. Conclusions and Future Perspectives
Funding
Conflicts of Interest
Abbreviations
References
- Macias, R.I.; Armengol, C.; Marin, J.J. Hepatoblastoma etiopathogenesis. J. Carcinog. Mutagen. 2016, 7, 1–4. [Google Scholar]
- Meyers, R.L.; Maibach, R.; Hiyama, E.; Haberle, B.; Krailo, M.; Rangaswami, A.; Aronson, D.C.; Malogolowkin, M.H.; Perilongo, G.; von Schweinitz, D.; et al. Risk-stratified staging in paediatric hepatoblastoma: A unified analysis from the Children’s Hepatic tumors International Collaboration. Lancet Oncol. 2017, 18, 122–131. [Google Scholar] [CrossRef]
- Aronson, D.C.; Czauderna, P.; Maibach, R.; Perilongo, G.; Morland, B. The treatment of hepatoblastoma: Its evolution and the current status as per the SIOPEL trials. J. Indian Assoc. Pediatr. Surg. 2014, 19, 201–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czauderna, P.; Garnier, H. Hepatoblastoma: Current understanding, recent advances, and controversies. F1000Research 2018, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- De Ioris, M.; Brugieres, L.; Zimmermann, A.; Keeling, J.; Brock, P.; Maibach, R.; Pritchard, J.; Shafford, L.; Zsiros, J.; Czaudzerna, P.; et al. Hepatoblastoma with a low serum alpha-fetoprotein level at diagnosis: The SIOPEL group experience. Eur. J. Cancer 2008, 44, 545–550. [Google Scholar] [CrossRef]
- Haas, J.E.; Muczynski, K.A.; Krailo, M.; Ablin, A.; Land, V.; Vietti, T.J.; Hammond, G.D. Histopathology and prognosis in childhood hepatoblastoma and hepatocarcnoma. Cancer 1989, 64, 1082–1095. [Google Scholar]
- Agarwala, S.; Gupta, A.; Bansal, D.; Vora, T.; Prasad, M.; Arora, B.; Kapoor, G.; Chinnaswamy, G.; Radhakrishnan, V.; Laskar, S.; et al. Management of Hepatoblastoma: ICMR Consensus Document. Indian J. Pediatr. 2017, 84, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh-Lotrario, A.D.; Tomlinson, G.E.; Finegold, M.J.; Gore, L.; Feusner, J.H. Small cell undifferentiated variant of hepatoblastoma: Adverse clinical and molecular features similar to rhabdoid tumors. Pediatr. Blood Cancer 2009, 52, 328–334. [Google Scholar] [CrossRef]
- Hooks, K.B.; Audoux, J.; Fazli, H.; Lesjean, S.; Ernault, T.; Dugot-Senant, N.; Leste-Lasserre, T.; Hagedorn, M.; Rousseau, B.; Danet, C.; et al. New insights into diagnosis and therapeutic options for proliferative hepatoblastoma. Hepatology 2018, 68, 89–102. [Google Scholar] [CrossRef]
- Asensio, M.; Lozano, E.; Cives-Losada, C.; Carrillo, J.; Abete, L.; Briz, O.; Al-Abdulla, R.; Alonso-Peña, M.; Perez-Silva, L.; Cairo, S.; et al. Role of transportome in chemoresistance of hepatoblastoma. J. Physiol. Biochem. 2018, 74, S90. [Google Scholar]
- Marin, J.J.G.; Briz, O.; Herraez, E.; Lozano, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Osorio-Padilla, L.M.; Santos-Llamas, A.I.; Serrano, M.A.; et al. Molecular bases of the poor response of liver cancer to chemotherapy. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.; Briz, O.; Monte, M.J.; Blazquez, A.G.; Macias, R.I. Genetic variants in genes involved in mechanisms of chemoresistance to anticancer drugs. Curr. Cancer Drug Targets 2012, 12, 402–438. [Google Scholar] [CrossRef] [PubMed]
- Muggia, F.M.; Bonetti, A.; Hoeschele, J.D.; Rozencweig, M.; Howell, S.B. Platinum Antitumor Complexes: 50 Years Since Barnett Rosenberg’s Discovery. J. Clin. Oncol. 2015, 33, 4219–4226. [Google Scholar] [CrossRef] [PubMed]
- Gatti, L.; Cassinelli, G.; Zaffaroni, N.; Lanzi, C.; Perego, P. New mechanisms for old drugs: Insights into DNA-unrelated effects of platinum compounds and drug resistance determinants. Drug Resist. Update 2015, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Masuda, S.; Yokoo, S.; Katsura, T.; Inui, K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family). J. Pharmacol. Exp. Ther. 2006, 319, 879–886. [Google Scholar] [CrossRef]
- Guttmann, S.; Chandhok, G.; Groba, S.R.; Niemietz, C.; Sauer, V.; Gomes, A.; Ciarimboli, G.; Karst, U.; Zibert, A.; Schmidt, H.H. Organic cation transporter 3 mediates cisplatin and copper cross-resistance in hepatoma cells. Oncotarget 2018, 9, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, C.S.; Sprowl, J.A.; Walker, A.L.; Hu, S.; Gibson, A.A.; Sparreboom, A. Modulation of OATP1B-type transporter function alters cellular uptake and disposition of platinum chemotherapeutics. Mol. Cancer Ther. 2013, 12, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Briz, O.; Serrano, M.A.; Rebollo, N.; Hagenbuch, B.; Meier, P.J.; Koepsell, H.; Marin, J.J. Carriers involved in targeting the cytostatic bile acid-cisplatin derivatives cis-diammine-chloro-cholylglycinate-platinum(II) and cis-diammine-bisursodeoxycholate-platinum(II) toward liver cells. Mol. Pharmacol. 2002, 61, 853–860. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Kobayashi, M.; Okada, M.; Takeuchi, T.; Unno, M.; Abe, T.; Goto, J.; Hishinuma, T.; Mano, N. Rapid screening of antineoplastic candidates for the human organic anion transporter OATP1B3 substrates using fluorescent probes. Cancer Lett. 2008, 260, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzer, A.K.; Manorek, G.H.; Howell, S.B. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol. Pharmacol. 2006, 70, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Larson, C.A.; Blair, B.G.; Safaei, R.; Howell, S.B. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Mol. Pharmacol. 2009, 75, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Holzer, A.K.; Katano, K.; Klomp, L.W.; Howell, S.B. Cisplatin rapidly down-regulates its own influx transporter hCTR1 in cultured human ovarian carcinoma cells. Clin. Cancer Res. 2004, 10, 6744–6749. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.; Monte, M.J.; Blazquez, A.G.; Macias, R.I.; Serrano, M.A.; Briz, O. The role of reduced intracellular concentrations of active drugs in the lack of response to anticancer chemotherapy. Acta. Pharmacol. Sin. 2014, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ruck, P.; Xiao, J.C. Stem-like cells in hepatoblastoma. Med. Pediatr. Oncol. 2002, 39, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Warmann, S.; Hunger, M.; Teichmann, B.; Flemming, P.; Gratz, K.F.; Fuchs, J. The role of the MDR1 gene in the development of multidrug resistance in human hepatoblastoma: Clinical course and in vivo model. Cancer 2002, 95, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Oue, T.; Yoneda, A.; Uehara, S.; Yamanaka, H.; Fukuzawa, M. Increased expression of multidrug resistance-associated genes after chemotherapy in pediatric solid malignancies. J. Pediatr. Surg. 2009, 44, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Warmann, S.W.; Heitmann, H.; Teichmann, B.; Gratz, K.F.; Ruck, P.; Hunger, M.; Fuchs, J. Effects of P-glycoprotein modulation on the chemotherapy of xenotransplanted human hepatoblastoma. Pediatr. Hematol. Oncol. 2005, 22, 373–386. [Google Scholar] [CrossRef]
- Vander Borght, S.; van Pelt, J.; van Malenstein, H.; Cassiman, D.; Renard, M.; Verslype, C.; Libbrecht, L.; Roskams, T.A. Up-regulation of breast cancer resistance protein expression in hepatoblastoma following chemotherapy: A study in patients and in vitro. Hepatol. Res. 2008, 38, 1112–1121. [Google Scholar] [CrossRef]
- Martinez-Becerra, P.; Vaquero, J.; Romero, M.R.; Lozano, E.; Anadon, C.; Macias, R.I.; Serrano, M.A.; Grane-Boladeras, N.; Munoz-Bellvis, L.; Alvarez, L.; et al. No correlation between the expression of FXR and genes involved in multidrug resistance phenotype of primary liver tumors. Mol. Pharm. 2012, 9, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Konig, J.; Buchholz, J.K.; Spring, H.; Leier, I.; Keppler, D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol. Pharmacol. 1999, 55, 929–937. [Google Scholar]
- Liu, X.Y.; Liu, S.P.; Jiang, J.; Zhang, X.; Zhang, T. Inhibition of the JNK signaling pathway increases sensitivity of hepatocellular carcinoma cells to cisplatin by down-regulating expression of P-glycoprotein. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 1098–1108. [Google Scholar] [PubMed]
- Korita, P.V.; Wakai, T.; Shirai, Y.; Matsuda, Y.; Sakata, J.; Takamura, M.; Yano, M.; Sanpei, A.; Aoyagi, Y.; Hatakeyama, K.; et al. Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with hepatocellular carcinoma. Oncol. Rep. 2010, 23, 965–972. [Google Scholar] [PubMed]
- Wakamatsu, T.; Nakahashi, Y.; Hachimine, D.; Seki, T.; Okazaki, K. The combination of glycyrrhizin and lamivudine can reverse the cisplatin resistance in hepatocellular carcinoma cells through inhibition of multidrug resistance-associated proteins. Int. J. Oncol. 2007, 31, 1465–1472. [Google Scholar] [CrossRef]
- Samimi, G.; Safaei, R.; Katano, K.; Holzer, A.K.; Rochdi, M.; Tomioka, M.; Goodman, M.; Howell, S.B. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin. Cancer Res. 2004, 10, 4661–4669. [Google Scholar] [CrossRef]
- Safaei, R.; Otani, S.; Larson, B.J.; Rasmussen, M.L.; Howell, S.B. Transport of cisplatin by the copper efflux transporter ATP7B. Mol. Pharmacol. 2008, 73, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Hu, X.; Xu, B.; Tong, T.; Jing, Y.; Xi, L.; Zhou, W.; Lu, J.; Wang, X.; Yang, X.; et al. Glutathione Stransferase isozyme alpha 1 is predominantly involved in the cisplatin resistance of common types of solid cancer. Oncol. Rep. 2019, 41, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Bader, P.; Fuchs, J.; Wenderoth, M.; von Schweinitz, D.; Niethammer, D.; Beck, J.F. Altered expression of resistance associated genes in hepatoblastoma xenografts incorporated into mice following treatment with adriamycin or cisplatin. Anticancer Res. 1998, 18, 3127–3132. [Google Scholar] [PubMed]
- Lieber, J.; Kirchner, B.; Eicher, C.; Warmann, S.W.; Seitz, G.; Fuchs, J.; Armeanu-Ebinger, S. Inhibition of Bcl-2 and Bcl-X enhances chemotherapy sensitivity in hepatoblastoma cells. Pediatr. Blood Cancer 2010, 55, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Lieber, J.; Ellerkamp, V.; Wenz, J.; Kirchner, B.; Warmann, S.W.; Fuchs, J.; Armeanu-Ebinger, S. Apoptosis sensitizers enhance cytotoxicity in hepatoblastoma cells. Pediatr. Surg. Int. 2012, 28, 149–159. [Google Scholar] [CrossRef]
- Lieber, J.; Dewerth, A.; Wenz, J.; Kirchner, B.; Eicher, C.; Warmann, S.W.; Fuchs, J.; Armeanu-Ebinger, S. Increased efficacy of CDDP in a xenograft model of hepatoblastoma using the apoptosis sensitizer ABT-737. Oncol. Rep. 2013, 29, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhao, X.; Zhang, Y.; Kang, Y.; Wang, J.; Liu, Y. Antitumor activity of YM155, a selective survivin suppressant, in combination with cisplatin in hepatoblastoma. Oncol. Rep. 2015, 34, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Q.; Wu, X.; Gan, L.; Yang, X.L.; Miao, Z.H. Hypoxia induces universal but differential drug resistance and impairs anticancer mechanisms of 5-fluorouracil in hepatoma cells. Acta. Pharmacol. Sin. 2017, 38, 1642–1654. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Chen, X.; Chen, S.; Ye, W.; Hou, K.; Liang, M. Arsenic trioxide reduces chemo-resistance to 5-fluorouracil and cisplatin in HBx-HepG2 cells via complex mechanisms. Cancer Cell Int. 2015, 15, 116. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.J.; Feng, C.W.; Li, M. ADAM17 mediates hypoxia-induced drug resistance in hepatocellular carcinoma cells through activation of EGFR/PI3K/Akt pathway. Mol. Cell. Biochem. 2013, 380, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Brozovic, A. The relationship between platinum drug resistance and epithelial-mesenchymal transition. Arch. Toxicol. 2017, 91, 605–619. [Google Scholar] [CrossRef]
- Li, R.; Wu, C.; Liang, H.; Zhao, Y.; Lin, C.; Zhang, X.; Ye, C. Knockdown of TWIST enhances the cytotoxicity of chemotherapeutic drugs in doxorubicin-resistant HepG2 cells by suppressing MDR1 and EMT. Int. J. Oncol. 2018, 53, 1763–1773. [Google Scholar] [CrossRef]
- Iv Santaliz-Ruiz, L.E.; Xie, X.; Old, M.; Teknos, T.N.; Pan, Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int. J. Cancer. 2014, 135, 2741–2748. [Google Scholar] [CrossRef]
- Yu, A.Q.; Ding, Y.; Li, C.L.; Yang, Y.; Yan, S.R.; Li, D.S. TALEN-induced disruption of Nanog expression results in reduced proliferation, invasiveness and migration, increased chemosensitivity and reversal of EMT in HepG2 cells. Oncol. Rep. 2016, 35, 1657–1663. [Google Scholar] [CrossRef]
- Adachi, T.; Nakagawa, H.; Chung, I.; Hagiya, Y.; Hoshijima, K.; Noguchi, N.; Kuo, M.T.; Ishikawa, T. Nrf2-dependent and -independent induction of ABC transporters ABCC1, ABCC2, and ABCG2 in HepG2 cells under oxidative stress. J. Exp. Ther. Oncol. 2007, 6, 335–348. [Google Scholar]
- Sumazin, P.; Chen, Y.; Trevino, L.R.; Sarabia, S.F.; Hampton, O.A.; Patel, K.; Mistretta, T.A.; Zorman, B.; Thompson, P.; Heczey, A.; et al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 2017, 65, 104–121. [Google Scholar] [CrossRef]
- Terashima, J.; Tachikawa, C.; Kudo, K.; Habano, W.; Ozawa, S. An aryl hydrocarbon receptor induces VEGF expression through ATF4 under glucose deprivation in HepG2. BMC Mol. Biol. 2013, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Malogolowkin, M.H.; Katzenstein, H.M.; Krailo, M.; Chen, Z.; Quinn, J.J.; Reynolds, M.; Ortega, J.A. Redefining the role of doxorubicin for the treatment of children with hepatoblastoma. J. Clin. Oncol. 2008, 26, 2379–2383. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, E.; Ueda, Y.; Onitake, Y.; Kurihara, S.; Watanabe, K.; Hishiki, T.; Tajiri, T.; Ida, K.; Yano, M.; Kondo, S.; et al. A cisplatin plus pirarubicin-based JPLT2 chemotherapy for hepatoblastoma: Experience and future of the Japanese Study Group for Pediatric Liver Tumor (JPLT). Pediatr. Surg. Int. 2013, 29, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Regev, R.; Yeheskely-Hayon, D.; Katzir, H.; Eytan, G.D. Transport of anthracyclines and mitoxantrone across membranes by a flip-flop mechanism. Biochem. Pharmacol. 2005, 70, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Andreev, E.; Brosseau, N.; Carmona, E.; Mes-Masson, A.M.; Ramotar, D. The human organic cation transporter OCT1 mediates high affinity uptake of the anticancer drug daunorubicin. Sci. Rep. 2016, 6, 20508. [Google Scholar] [CrossRef] [Green Version]
- Okabe, M.; Szakacs, G.; Reimers, M.A.; Suzuki, T.; Hall, M.D.; Abe, T.; Weinstein, J.N.; Gottesman, M.M. Profiling SLCO and SLC22 genes in the NCI-60 cancer cell lines to identify drug uptake transporters. Mol. Cancer Ther. 2008, 7, 3081–3091. [Google Scholar] [CrossRef] [Green Version]
- Okabe, M.; Unno, M.; Harigae, H.; Kaku, M.; Okitsu, Y.; Sasaki, T.; Mizoi, T.; Shiiba, K.; Takanaga, H.; Terasaki, T.; et al. Characterization of the organic cation transporter SLC22A16: A doxorubicin importer. Biochem. Biophys. Res. Commun. 2005, 333, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Herraez, E.; Lozano, E.; Macias, R.I.; Vaquero, J.; Bujanda, L.; Banales, J.M.; Marin, J.J.; Briz, O. Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 2013, 58, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Angelini, S.; Pantaleo, M.A.; Ravegnini, G.; Zenesini, C.; Cavrini, G.; Nannini, M.; Fumagalli, E.; Palassini, E.; Saponara, M.; Di Battista, M.; et al. Polymorphisms in OCTN1 and OCTN2 transporters genes are associated with prolonged time to progression in unresectable gastrointestinal stromal tumours treated with imatinib therapy. Pharmacol. Res. 2013, 68, 1–6. [Google Scholar] [CrossRef]
- Chen, Y.B.; Yan, M.L.; Gong, J.P.; Xia, R.P.; Liu, L.X.; Li, N.; Lu, S.C.; Zhang, J.G.; Zeng, D.B.; Xie, J.G.; et al. Establishment of hepatocellular carcinoma multidrug resistant monoclone cell line HepG2/mdr1. Chin. Med. J. (Engl.) 2007, 120, 703–707. [Google Scholar] [CrossRef]
- Jani, M.; Ambrus, C.; Magnan, R.; Jakab, K.T.; Beery, E.; Zolnerciks, J.K.; Krajcsi, P. Structure and function of BCRP, a broad specificity transporter of xenobiotics and endobiotics. Arch. Toxicol. 2014, 88, 1205–1248. [Google Scholar] [CrossRef] [PubMed]
- He, S.M.; Li, R.; Kanwar, J.R.; Zhou, S.F. Structural and functional properties of human multidrug resistance protein 1 (MRP1/ABCC1). Curr. Med. Chem. 2011, 18, 439–481. [Google Scholar] [CrossRef] [PubMed]
- Vander Borght, S.; Komuta, M.; Libbrecht, L.; Katoonizadeh, A.; Aerts, R.; Dymarkowski, S.; Verslype, C.; Nevens, F.; Roskams, T. Expression of multidrug resistance-associated protein 1 in hepatocellular carcinoma is associated with a more aggressive tumour phenotype and may reflect a progenitor cell origin. Liver Int. 2008, 28, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Eicher, C.; Dewerth, A.; Kirchner, B.; Warmann, S.W.; Fuchs, J.; Armeanu-Ebinger, S. Development of a drug resistance model for hepatoblastoma. Int. J. Oncol. 2011, 38, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.S.; Tiwari, A.K. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 2011, 278, 3226–3245. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Hu, B.; Tang, L.; Zheng, S.; Sun, Y.; Sheng, Z.; Yao, Y.; Lin, F. The overexpression of MRP4 is related to multidrug resistance in osteosarcoma cells. J. Cancer. Res. Ther. 2015, 11, 18–23. [Google Scholar] [CrossRef]
- Borst, P.; de Wolf, C.; van de Wetering, K. Multidrug resistance-associated proteins 3, 4, and 5. Pflugers Arch. 2007, 453, 661–673. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Chen, Z.S.; Shchaveleva, I.; Zeng, H.; Kruh, G.D. Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6). Cancer Res. 2002, 62, 6172–6177. [Google Scholar]
- Hopper-Borge, E.; Xu, X.; Shen, T.; Shi, Z.; Chen, Z.S.; Kruh, G.D. Human multidrug resistance protein 7 (ABCC10) is a resistance factor for nucleoside analogues and epothilone B. Cancer Res. 2009, 69, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Smuc, T.; Rizner, T.L. Expression of 17beta-hydroxysteroid dehydrogenases and other estrogen-metabolizing enzymes in different cancer cell lines. Chem. Biol. Interact. 2009, 178, 228–233. [Google Scholar] [CrossRef]
- Eichenmuller, M.; Trippel, F.; Kreuder, M.; Beck, A.; Schwarzmayr, T.; Haberle, B.; Cairo, S.; Leuschner, I.; von Schweinitz, D.; Strom, T.M.; et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J. Hepatol. 2014, 61, 1312–1320. [Google Scholar] [CrossRef]
- Tashiro, K.; Asakura, T.; Fujiwara, C.; Ohkawa, K.; Ishibashi, Y. Glutathione-S-transferase-pi expression regulates sensitivity to glutathione-doxorubicin conjugate. Anticancer Drugs 2001, 12, 707–712. [Google Scholar] [CrossRef]
- Ganapathi, R.N.; Ganapathi, M.K. Mechanisms regulating resistance to inhibitors of topoisomerase II. Front. Pharmacol. 2013, 4, 89. [Google Scholar] [CrossRef]
- Spencer, D.M.; Bilardi, R.A.; Koch, T.H.; Post, G.C.; Nafie, J.W.; Kimura, K.; Cutts, S.M.; Phillips, D.R. DNA repair in response to anthracycline-DNA adducts: A role for both homologous recombination and nucleotide excision repair. Mutat. Res. 2008, 638, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, N.; Ye, Y.; Qian, J.; Zhu, Y.; Wang, C. Role and mechanisms of microRNA503 in drug resistance reversal in HepG2/ADM human hepatocellular carcinoma cells. Mol. Med. Rep. 2014, 10, 3268–3274. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Lau, T.C.; Ng, I.O. Doxorubicin-induced apoptosis and chemosensitivity in hepatoma cell lines. Cancer Chemother. Pharmacol. 2002, 49, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Soini, T.; Pihlajoki, M.; Kyronlahti, A.; Andersson, L.C.; Wilson, D.B.; Heikinheimo, M. Downregulation of transcription factor GATA4 sensitizes human hepatoblastoma cells to doxorubicin-induced apoptosis. Tumour Biol. 2017, 39, 1010428317695016. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Cheng, S.C.; Xie, H.; Xie, Y. Effects of Bcl-2 and Bcl-XL protein levels on chemoresistance of hepatoblastoma HepG2 cell line. Biochem. Cell Biol. 2000, 78, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Warmann, S.W.; Frank, H.; Heitmann, H.; Ruck, P.; Herberts, T.; Seitz, G.; Fuchs, J. Bcl-2 gene silencing in pediatric epithelial liver tumors. J. Surg. Res. 2008, 144, 43–48. [Google Scholar] [CrossRef]
- Jin, J.; Woodfield, S.E.; Patel, R.H.; Jin, N.G.; Shi, Y.; Liu, B.; Sun, W.; Chen, X.; Yu, Y.; Vasudevan, S.A. Targeting LRH1 in hepatoblastoma cell lines causes decreased proliferation. Oncol. Rep. 2019, 41, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.C.; Chen, P.H.; Cheng, C.I.; Tsai, M.S.; Chang, C.Y.; Lu, S.C.; Hsieh, M.C.; Lin, Y.C.; Lee, P.H.; Kao, Y.H. Toll-like receptor-4 is a target for suppression of proliferation and chemoresistance in HepG2 hepatoblastoma cells. Cancer Lett. 2015, 368, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Purcell, R.; Childs, M.; Maibach, R.; Miles, C.; Turner, C.; Zimmermann, A.; Sullivan, M. HGF/c-Met related activation of beta-catenin in hepatoblastoma. J. Exp. Clin. Cancer Res. 2011, 30, 96. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, C.; Lewis, A.L.; Lloyd, A.W.; Phillips, G.J.; Macfarlane, W.M. Hypoxia as a target for drug combination therapy of liver cancer. Anticancer Drugs 2017, 28, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.D.; Chen, W.; Yan, T.L.; Ma, T.; Chen, C.L.; Liang, C.; Zhang, Q.; Xia, X.F.; Liu, H.; Zhi, X.; et al. NSC 74859 enhances doxorubicin cytotoxicity via inhibition of epithelial-mesenchymal transition in hepatocellular carcinoma cells. Cancer Lett. 2012, 325, 207–213. [Google Scholar] [CrossRef]
- Casanova, M.; Massimino, M.; Ferrari, A.; Spreafico, F.; Piva, L.; Coppa, J.; Luksch, R.; Cefalo, G.; Terenziani, M.; Polastri, D.; et al. Etoposide, cisplatin, epirubicin chemotherapy in the treatment of pediatric liver tumors. Pediatr. Hematol. Oncol. 2005, 22, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.A.; Douglass, E.C.; Feusner, J.H.; Reynolds, M.; Quinn, J.J.; Finegold, M.J.; Haas, J.E.; King, D.R.; Liu-Mares, W.; Sensel, M.G.; et al. Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children’s Cancer Group and the Pediatric Oncology Group. J. Clin. Oncol. 2000, 18, 2665–2675. [Google Scholar] [CrossRef]
- Fuchs, J.; Bode, U.; von Schweinitz, D.; Weinel, P.; Erttmann, R.; Harms, D.; Mildenberger, H. Analysis of treatment efficiency of carboplatin and etoposide in combination with radical surgery in advanced and recurrent childhood hepatoblastoma: A report of the German Cooperative Pediatric Liver Tumor Study HB 89 and HB 94. Klin. Padiatr. 1999, 211, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Lancaster, C.S.; Zuo, Z.; Hu, S.; Chen, Z.; Rubnitz, J.E.; Baker, S.D.; Sparreboom, A. Inhibition of OCTN2-mediated transport of carnitine by etoposide. Mol. Cancer Ther. 2012, 11, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Marada, V.V.; Florl, S.; Kuhne, A.; Burckhardt, G.; Hagos, Y. Interaction of human organic anion transporter polypeptides 1B1 and 1B3 with antineoplastic compounds. Eur. J. Med. Chem. 2015, 92, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Lagas, J.S.; Fan, L.; Wagenaar, E.; Vlaming, M.L.; van Tellingen, O.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein (P-gp/Abcb1), Abcc2, and Abcc3 determine the pharmacokinetics of etoposide. Clin. Cancer Res. 2010, 16, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Kawashiro, T.; Yamashita, K.; Zhao, X.J.; Koyama, E.; Tani, M.; Chiba, K.; Ishizaki, T. A study on the metabolism of etoposide and possible interactions with antitumor or supporting agents by human liver microsomes. J. Pharmacol. Exp. Ther. 1998, 286, 1294–1300. [Google Scholar] [PubMed]
- Sumida, A.; Yamamoto, I.; Zhou, Q.; Morisaki, T.; Azuma, J. Evaluation of induction of CYP3A mRNA using the HepG2 cell line and reverse transcription-PCR. Biol. Pharm. Bull. 1999, 22, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.G.; Mackenzie, P.I.; Lu, L.; Meech, R.; McKinnon, R.A. Induction of human UDP-Glucuronosyltransferase 2B7 gene expression by cytotoxic anticancer drugs in liver cancer HepG2 cells. Drug Metab. Dispos. 2015, 43, 660–668. [Google Scholar] [CrossRef]
- Burden, D.A.; Kingma, P.S.; Froelich-Ammon, S.J.; Bjornsti, M.A.; Patchan, M.W.; Thompson, R.B.; Osheroff, N. Topoisomerase II.etoposide interactions direct the formation of drug-induced enzyme-DNA cleavage complexes. J. Biol. Chem. 1996, 271, 29238–29244. [Google Scholar] [CrossRef] [PubMed]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006, 407, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Eicher, C.; Dewerth, A.; Kirchner, B.; Warmann, S.W.; Fuchs, J.; Armeanu-Ebinger, S. Treatment effects of the multikinase inhibitor sorafenib on hepatoblastoma cell lines and xenografts in NMRI-Foxn1 nu mice. Liver Int. 2012, 32, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Eicher, C.; Dewerth, A.; Thomale, J.; Ellerkamp, V.; Hildenbrand, S.; Warmann, S.W.; Fuchs, J.; Armeanu-Ebinger, S. Effect of sorafenib combined with cytostatic agents on hepatoblastoma cell lines and xenografts. Br. J. Cancer. 2013, 108, 334–341. [Google Scholar] [CrossRef]
- Schmid, I.; Haberle, B.; Albert, M.H.; Corbacioglu, S.; Frohlich, B.; Graf, N.; Kammer, B.; Kontny, U.; Leuschner, I.; Scheel-Walter, H.G.; et al. Sorafenib and cisplatin/doxorubicin (PLADO) in pediatric hepatocellular carcinoma. Pediatr. Blood Cancer 2012, 58, 539–544. [Google Scholar] [CrossRef]
- Al-Abdulla, R.; Lozano, E.; Macias, R.I.R.; Monte, M.J.; Briz, O.; O’Rourke, C.J.; Serrano, M.A.; Banales, J.M.; Avila, M.A.; Martinez-Chantar, M.L.; et al. Epigenetic events involved in OCT1-dependent impaired response of hepatocellular carcinoma to sorafenib. Br. J. Pharmacol. 2019, 176, 787–800. [Google Scholar] [CrossRef]
- Lozano, E.; Macias, R.I.; Monte, M.J.; Asensio, M.; del Carmen, S.; Sanchez-Vicente, L.; Alonso-Peña, M.; Al-Abdulla, R.; O’Rourke, C.J.; Banales, J.M.; et al. Genetic and epigenetic events in the regulation of the expression of the organic cation transporter OCT1: Role in chemoresistance of liver cancer to sorafenib. J. Physiol. Biochem. 2018, 74, S46–S47. [Google Scholar]
- Geier, A.; Macias, R.I.; Bettinger, D.; Weiss, J.; Bantel, H.; Jahn, D.; Al-Abdulla, R.; Marin, J.J. The lack of the organic cation transporter OCT1 at the plasma membrane of tumor cells precludes a positive response to sorafenib in patients with hepatocellular carcinoma. Oncotarget 2017, 8, 15846–15857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haouala, A.; Rumpold, H.; Untergasser, G.; Buclin, T.; Ris, H.B.; Widmer, N.; Decosterd, L.A. siRNA-mediated knock-down of P-glycoprotein expression reveals distinct cellular disposition of anticancer tyrosine kinases inhibitors. Drug Metab. Lett. 2010, 4, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Lagas, J.S.; van Waterschoot, R.A.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol. Cancer Ther. 2010, 9, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Hsieh, Y.L.; Hung, C.M.; Chien, P.H.; Chien, Y.F.; Chen, L.C.; Tu, C.Y.; Chen, C.H.; Hsu, S.C.; Lin, Y.M.; et al. BCRP/ABCG2 inhibition sensitizes hepatocellular carcinoma cells to sorafenib. PLoS ONE 2013, 8, e83627. [Google Scholar] [CrossRef] [PubMed]
- Tandia, M.; Mhiri, A.; Paule, B.; Saffroy, R.; Cailliez, V.; Noe, G.; Farinotti, R.; Bonhomme-Faivre, L. Correlation between clinical response to sorafenib in hepatocellular carcinoma treatment and polymorphisms of P-glycoprotein (ABCB1) and of breast cancer resistance protein (ABCG2): Monocentric study. Cancer Chemother. Pharmacol. 2017, 79, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, Y.; Nakano, K.; Maeda, H.; Taguchi, M.; Ikeda, R.; Sugawara, M.; Iseki, K.; Takeda, Y.; Yamada, K. Multidrug resistance protein 2 implicates anticancer drug-resistance to sorafenib. Biol. Pharm. Bull. 2011, 34, 433–435. [Google Scholar] [CrossRef]
- Narjoz, C.; Favre, A.; McMullen, J.; Kiehl, P.; Montemurro, M.; Figg, W.D.; Beaune, P.; de Waziers, I.; Rochat, B. Important role of CYP2J2 in protein kinase inhibitor degradation: A possible role in intratumor drug disposition and resistance. PLoS ONE 2014, 9, e95532. [Google Scholar] [CrossRef]
- Ghassabian, S.; Rawling, T.; Zhou, F.; Doddareddy, M.R.; Tattam, B.N.; Hibbs, D.E.; Edwards, R.J.; Cui, P.H.; Murray, M. Role of human CYP3A4 in the biotransformation of sorafenib to its major oxidized metabolites. Biochem. Pharmacol. 2012, 84, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Scartozzi, M.; Faloppi, L.; Svegliati Baroni, G.; Loretelli, C.; Piscaglia, F.; Iavarone, M.; Toniutto, P.; Fava, G.; De Minicis, S.; Mandolesi, A.; et al. VEGF and VEGFR genotyping in the prediction of clinical outcome for HCC patients receiving sorafenib: The ALICE-1 study. Int. J. Cancer 2014, 135, 1247–1256. [Google Scholar] [CrossRef] [Green Version]
- Macias, R.I.R.; Sanchez-Martin, A.; Rodriguez-Macias, G.; Sanchez-Abarca, L.I.; Lozano, E.; Herraez, E.; Odero, M.D.; Diez-Martin, J.L.; Marin, J.J.G.; Briz, O. Role of drug transporters in the sensitivity of acute myeloid leukemia to sorafenib. Oncotarget 2018, 9, 28474–28485. [Google Scholar] [CrossRef]
- Negri, F.V.; Dal Bello, B.; Porta, C.; Campanini, N.; Rossi, S.; Tinelli, C.; Poggi, G.; Missale, G.; Fanello, S.; Salvagni, S.; et al. Expression of pERK and VEGFR-2 in advanced hepatocellular carcinoma and resistance to sorafenib treatment. Liver Int. 2015, 35, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, A.; Navarro-Villaran, E.; Gonzalez, R.; Pereira, S.; Soriano-De Castro, L.B.; Sarrias-Gimenez, A.; Barrera-Pulido, L.; Alamo-Martinez, J.M.; Serrablo-Requejo, A.; Blanco-Fernandez, G.; et al. Regulation of cell death receptor S-nitrosylation and apoptotic signaling by Sorafenib in hepatoblastoma cells. Redox Biol. 2015, 6, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Tutusaus, A.; Stefanovic, M.; Boix, L.; Cucarull, B.; Zamora, A.; Blasco, L.; de Frutos, P.G.; Reig, M.; Fernandez-Checa, J.C.; Mari, M.; et al. Antiapoptotic BCL-2 proteins determine sorafenib/regorafenib resistance and BH3-mimetic efficacy in hepatocellular carcinoma. Oncotarget 2018, 9, 16701–16717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Zhai, B.; Sun, W.; Hu, F.; Cheng, H.; Xu, J. Activation of phosphatidylinositol 3-kinase/AKT/snail signaling pathway contributes to epithelial-mesenchymal transition-induced multi-drug resistance to sorafenib in hepatocellular carcinoma cells. PLoS ONE 2017, 12, e0185088. [Google Scholar] [CrossRef]
- Nagel, C.; Armeanu-Ebinger, S.; Dewerth, A.; Warmann, S.W.; Fuchs, J. Anti-tumor activity of sorafenib in a model of a pediatric hepatocellular carcinoma. Exp. Cell Res. 2015, 331, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K. Current chemotherapeutic approaches for hepatoblastoma. Int. J. Clin. Oncol. 2013, 18, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, J.; Thevelin, L.; Bing, Q.; Stieger, B.; Chanteux, H.; Augustijns, P.; Annaert, P. Role of the OATP Transporter Family and a Benzbromarone-SensitiveEfflux Transporter in the Hepatocellular Disposition of Vincristine. Pharm. Res. 2017, 34, 2336–2348. [Google Scholar] [CrossRef] [PubMed]
- Shnitsar, V.; Eckardt, R.; Gupta, S.; Grottker, J.; Muller, G.A.; Koepsell, H.; Burckhardt, G.; Hagos, Y. Expression of human organic cation transporter 3 in kidney carcinoma cell lines increases chemosensitivity to melphalan, irinotecan, and vincristine. Cancer Res. 2009, 69, 1494–1501. [Google Scholar] [CrossRef]
- Dumontet, C.; Sikic, B.I. Mechanisms of action of and resistance to antitubulin agents: Microtubule dynamics, drug transport, and cell death. J. Clin. Oncol. 1999, 17, 1061–1070. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, S.H.; Guo, X.L. New insights into Vinca alkaloids resistance mechanism and circumvention in lung cancer. Biomed. Pharmacother. 2017, 96, 659–666. [Google Scholar] [CrossRef]
- Chan, J.Y.; Chu, A.C.; Fung, K.P. Inhibition of P-glycoprotein expression and reversal of drug resistance of human hepatoma HepG2 cells by multidrug resistance gene (mdr1) antisense RNA. Life Sci. 2000, 67, 2117–2124. [Google Scholar] [CrossRef]
- Folmer, Y.; Schneider, M.; Blum, H.E.; Hafkemeyer, P. Reversal of drug resistance of hepatocellular carcinoma cells by adenoviral delivery of anti-ABCC2 antisense constructs. Cancer Gene Ther. 2007, 14, 875–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Trobaugh-Lotrario, A.D.; Katzenstein, H.M. Chemotherapeutic approaches for newly diagnosed hepatoblastoma: Past, present, and future strategies. Pediatr. Blood Cancer 2012, 59, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Valdes, R.; Casado, F.J.; Pastor-Anglada, M. Cell-cycle-dependent regulation of CNT1, a concentrative nucleoside transporter involved in the uptake of cell-cycle-dependent nucleoside-derived anticancer drugs. Biochem. Biophys. Res. Commun. 2002, 296, 575–579. [Google Scholar] [CrossRef]
- Zembutsu, H.; Ohnishi, Y.; Tsunoda, T.; Furukawa, Y.; Katagiri, T.; Ueyama, Y.; Tamaoki, N.; Nomura, T.; Kitahara, O.; Yanagawa, R.; et al. Genome-wide cDNA microarray screening to correlate gene expression profiles with sensitivity of 85 human cancer xenografts to anticancer drugs. Cancer Res. 2002, 62, 518–527. [Google Scholar]
- Wang, W.B.; Yang, Y.; Zhao, Y.P.; Zhang, T.P.; Liao, Q.; Shu, H. Recent studies of 5-fluorouracil resistance in pancreatic cancer. World J. Gastroenterol. 2014, 20, 15682–15690. [Google Scholar] [CrossRef]
- Pratt, S.; Shepard, R.L.; Kandasamy, R.A.; Johnston, P.A.; Perry, W., 3rd; Dantzig, A.H. The multidrug resistance protein 5 (ABCC5) confers resistance to 5-fluorouracil and transports its monophosphorylated metabolites. Mol. Cancer Ther. 2005, 4, 855–863. [Google Scholar] [CrossRef] [Green Version]
- Parker, W.B.; Cheng, Y.C. Metabolism and mechanism of action of 5-fluorouracil. Pharmacol. Ther. 1990, 48, 381–395. [Google Scholar] [CrossRef]
- Marin, J.J.; Al-Abdulla, R.; Lozano, E.; Briz, O.; Bujanda, L.; Banales, J.M.; Macias, R.I. Mechanisms of Resistance to Chemotherapy in Gastric Cancer. Anticancer Agents Med. Chem. 2016, 16, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Ozaki, T.; Nakanishi, M.; Kikuchi, H.; Yoshida, K.; Horie, H.; Kuwano, H.; Nakagawara, A. Oxidative stress induces p53-dependent apoptosis in hepatoblastoma cell through its nuclear translocation. Genes Cells 2007, 12, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Sarper, N.; Corapcioglu, F.; Anik, Y.; Ural, D.; Yildiz, K.; Tugay, M. Unresectable multifocal hepatoblastoma with cardiac extension: Excellent response with HB-94 chemotherapy protocol. J. Pediatr. Hematol. Oncol. 2006, 28, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, E.; Hishiki, T.; Watanabe, K.; Ida, K.; Yano, M.; Oue, T.; Iehara, T.; Hoshino, K.; Koh, K.; Tanaka, Y.; et al. Resectability and tumor response after preoperative chemotherapy in hepatoblastoma treated by the Japanese Study Group for Pediatric Liver Tumor (JPLT)-2 protocol. J. Pediatr. Surg. 2016, 51, 2053–2057. [Google Scholar] [CrossRef] [PubMed]
- Cacciavillano, W.D.; Brugieres, L.; Childs, M.; Shafford, E.; Brock, P.; Pritchard, J.; Mailbach, R.; Scopinaro, M.; Perilongo, G. Phase II study of high-dose cyclophosphamide in relapsing and/or resistant hepatoblastoma in children: A study from the SIOPEL group. Eur. J. Cancer 2004, 40, 2274–2279. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Holle, S.K.; Vollenbrocker, B.; Hagos, Y.; Reuter, S.; Burckhardt, G.; Bierer, S.; Herrmann, E.; Pavenstadt, H.; Rossi, R.; et al. New clues for nephrotoxicity induced by ifosfamide: Preferential renal uptake via the human organic cation transporter 2. Mol. Pharm. 2011, 8, 270–279. [Google Scholar] [CrossRef]
- Filipits, M.; Pohl, G.; Rudas, M.; Dietze, O.; Lax, S.; Grill, R.; Pirker, R.; Zielinski, C.C.; Hausmaninger, H.; Kubista, E.; et al. Clinical role of multidrug resistance protein 1 expression in chemotherapy resistance in early-stage breast cancer: The Austrian Breast and Colorectal Cancer Study Group. J. Clin. Oncol. 2005, 23, 1161–1168. [Google Scholar] [CrossRef]
- Tian, Q.; Zhang, J.; Tan, T.M.; Chan, E.; Duan, W.; Chan, S.Y.; Boelsterli, U.A.; Ho, P.C.; Yang, H.; Bian, J.S.; et al. Human multidrug resistance associated protein 4 confers resistance to camptothecins. Pharm. Res. 2005, 22, 1837–1853. [Google Scholar] [CrossRef]
- Zhang, J.; Ng, K.Y.; Ho, P.C. Interaction of oxazaphosphorines with multidrug resistance-associated protein 4 (MRP4). AAPS J. 2010, 12, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shen, Z.Y.; Xu, W.; Fan, T.Y.; Li, J.; Lu, Y.F.; Cheng, M.L.; Liu, J. Expression of P450 and nuclear receptors in normal and end-stage Chinese livers. World J. Gastroenterol. 2014, 20, 8681–8690. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Zhou, P.; Gupta, A.; Shin, S. Reactive Ductules Are Associated with Angiogenesis and Tumor Cell Proliferation in Pediatric Liver Cancer. Hepatol. Commun. 2018, 2, 1199–1212. [Google Scholar] [CrossRef] [PubMed]
- Marsh, A.M.; Lo, L.; Cohen, R.A.; Feusner, J.H. Sorafenib and bevacizumab for recurrent metastatic hepatoblastoma: Stable radiographic disease with decreased AFP. Pediatr. Blood Cancer 2012, 59, 939–940. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.F.; Oo, K.Z.; Fireman, F.; Pierre, L.; Bania, M.A.; Sadanandan, S.; Yamashiro, D.J.; Glade Bender, J.L. Reversible posterior leukoencephalopathy syndrome in a child treated with bevacizumab. Pediatr. Blood Cancer 2009, 52, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Arao, T.; Matsumoto, K.; Kimura, H.; Togashi, Y.; Hirashima, Y.; Horita, Y.; Iwasa, S.; Okita, N.T.; Honma, Y.; et al. Biomarkers of reactive resistance and early disease progression during chemotherapy plus bevacizumab treatment for colorectal carcinoma. Oncotarget 2014, 5, 2588–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, M.; Urabe, Y.; Ono, A.; Miki, D.; Ochi, H.; Chayama, K. Serial profiling of circulating tumor DNA for optimization of anti-VEGF chemotherapy in metastatic colorectal cancer patients. Int. J. Cancer 2018, 142, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kim, K.W.; Lee, Y.S.; Baek, J.H.; Kim, M.S.; Lee, Y.M.; Lee, M.S.; Kim, Y.J. Hypoglycemia-induced VEGF expression is mediated by intracellular Ca2+ and protein kinase C signaling pathway in HepG2 human hepatoblastoma cells. Int. J. Mol. Med. 2001, 7, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Kadenhe-Chiweshe, A.; Papa, J.; McCrudden, K.W.; Frischer, J.; Bae, J.O.; Huang, J.; Fisher, J.; Lefkowitch, J.H.; Feirt, N.; Rudge, J.; et al. Sustained VEGF blockade results in microenvironmental sequestration of VEGF by tumors and persistent VEGF receptor-2 activation. Mol. Cancer Res. 2008, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.M.; Pacheco, M.M.; Wickiser, J.E. Addition of Vincristine and Irinotecan to Standard Therapy in a Patient with Refractory High-risk Hepatoblastoma Achieving Long-term Relapse-free Survival. J. Pediatr. Hematol. Oncol. 2018. [Google Scholar] [CrossRef]
- O’Neill, A.F.; Towbin, A.J.; Krailo, M.D.; Xia, C.; Gao, Y.; McCarville, M.B.; Meyers, R.L.; McGahren, E.D.; Tiao, G.M.; Dunn, S.P.; et al. Characterization of Pulmonary Metastases in Children with Hepatoblastoma Treated on Children’s Oncology Group Protocol AHEP0731 (The Treatment of Children With All Stages of Hepatoblastoma): A Report From the Children’s Oncology Group. J. Clin. Oncol. 2017, 35, 3465–3473. [Google Scholar] [CrossRef]
- Gupta, S.; Wulf, G.; Henjakovic, M.; Koepsell, H.; Burckhardt, G.; Hagos, Y. Human organic cation transporter 1 is expressed in lymphoma cells and increases susceptibility to irinotecan and paclitaxel. J. Pharmacol. Exp. Ther. 2012, 341, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Hodges, L.M.; Markova, S.M.; Chinn, L.W.; Gow, J.M.; Kroetz, D.L.; Klein, T.E.; Altman, R.B. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharm. Genom. 2011, 21, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.F.; Pokharel, D.; Bebawy, M. MRP1 and its role in anticancer drug resistance. Drug. Metab. Rev. 2015, 47, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Mishra, L.; Banker, T.; Murray, J.; Byers, S.; Thenappan, A.; He, A.R.; Shetty, K.; Johnson, L.; Reddy, E.P. Liver stem cells and hepatocellular carcinoma. Hepatology 2009, 49, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Jeng, Y.M.; Wu, M.Z.; Mao, T.L.; Chang, M.H.; Hsu, H.C. Somatic mutations of beta-catenin play a crucial role in the tumorigenesis of sporadic hepatoblastoma. Cancer Lett. 2000, 152, 45–51. [Google Scholar] [CrossRef]
- Koch, A.; Denkhaus, D.; Albrecht, S.; Leuschner, I.; von Schweinitz, D.; Pietsch, T. Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res. 1999, 59, 269–273. [Google Scholar] [PubMed]
- Ranganathan, S.; Tan, X.; Monga, S.P. beta-Catenin and met deregulation in childhood Hepatoblastomas. Pediatr. Dev. Pathol. 2005, 8, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Hiyama, E.; Kamimatsuse, A.; Kamei, N.; Ogura, K.; Sueda, T. Wnt signaling and telomerase activation of hepatoblastoma: Correlation with chemosensitivity and surgical resectability. J. Pediatr. Surg. 2011, 46, 2221–2227. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marin, J.J.G.; Cives-Losada, C.; Asensio, M.; Lozano, E.; Briz, O.; Macias, R.I.R. Mechanisms of Anticancer Drug Resistance in Hepatoblastoma. Cancers 2019, 11, 407. https://doi.org/10.3390/cancers11030407
Marin JJG, Cives-Losada C, Asensio M, Lozano E, Briz O, Macias RIR. Mechanisms of Anticancer Drug Resistance in Hepatoblastoma. Cancers. 2019; 11(3):407. https://doi.org/10.3390/cancers11030407
Chicago/Turabian StyleMarin, Jose J. G., Candela Cives-Losada, Maitane Asensio, Elisa Lozano, Oscar Briz, and Rocio I. R. Macias. 2019. "Mechanisms of Anticancer Drug Resistance in Hepatoblastoma" Cancers 11, no. 3: 407. https://doi.org/10.3390/cancers11030407
APA StyleMarin, J. J. G., Cives-Losada, C., Asensio, M., Lozano, E., Briz, O., & Macias, R. I. R. (2019). Mechanisms of Anticancer Drug Resistance in Hepatoblastoma. Cancers, 11(3), 407. https://doi.org/10.3390/cancers11030407