Establishment and Characterization of a New Intrahepatic Cholangiocarcinoma Cell Line Resistant to Gemcitabine

 , ,
, , 
Abstract
:1. Introduction
2. Results
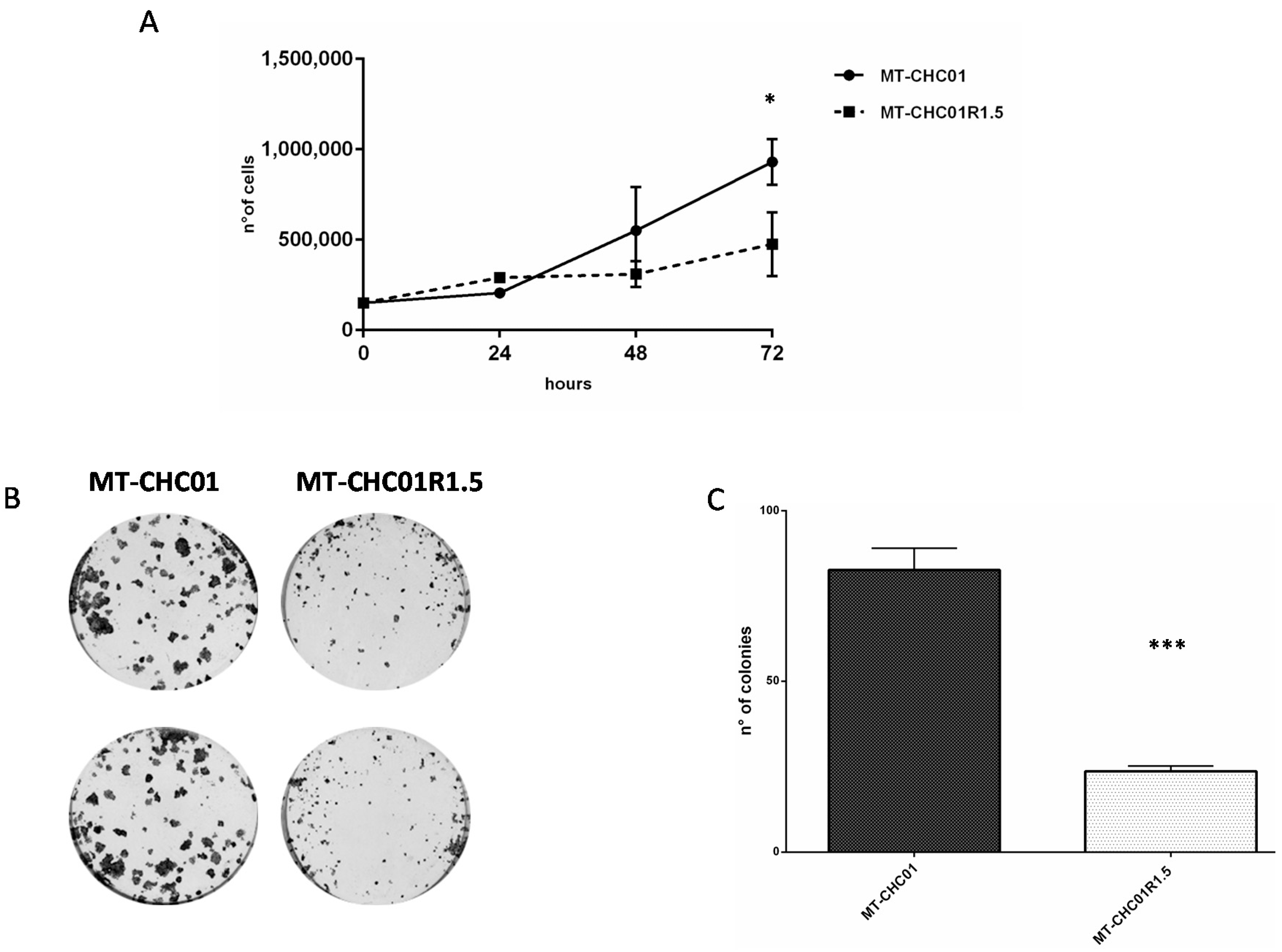
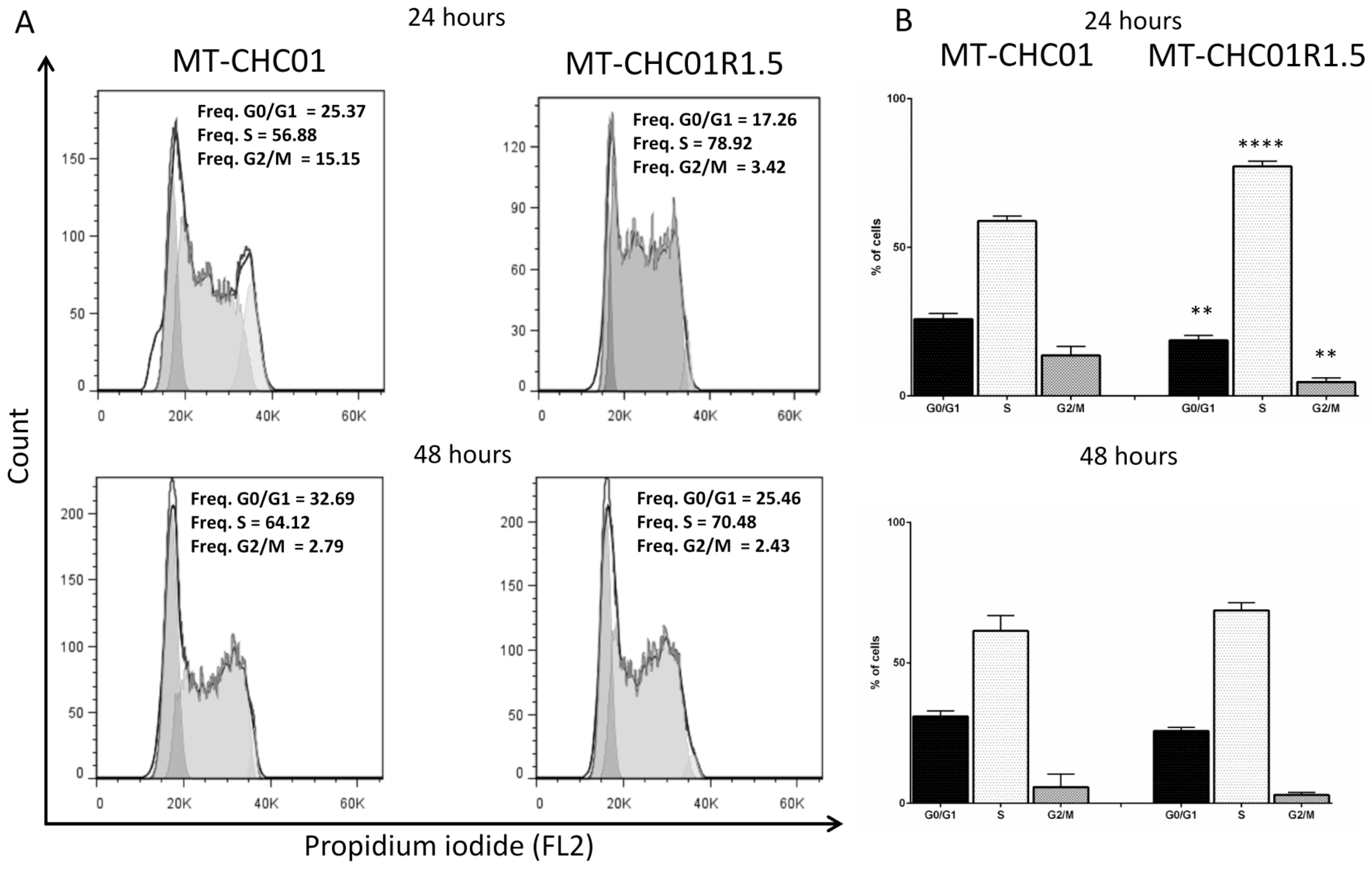
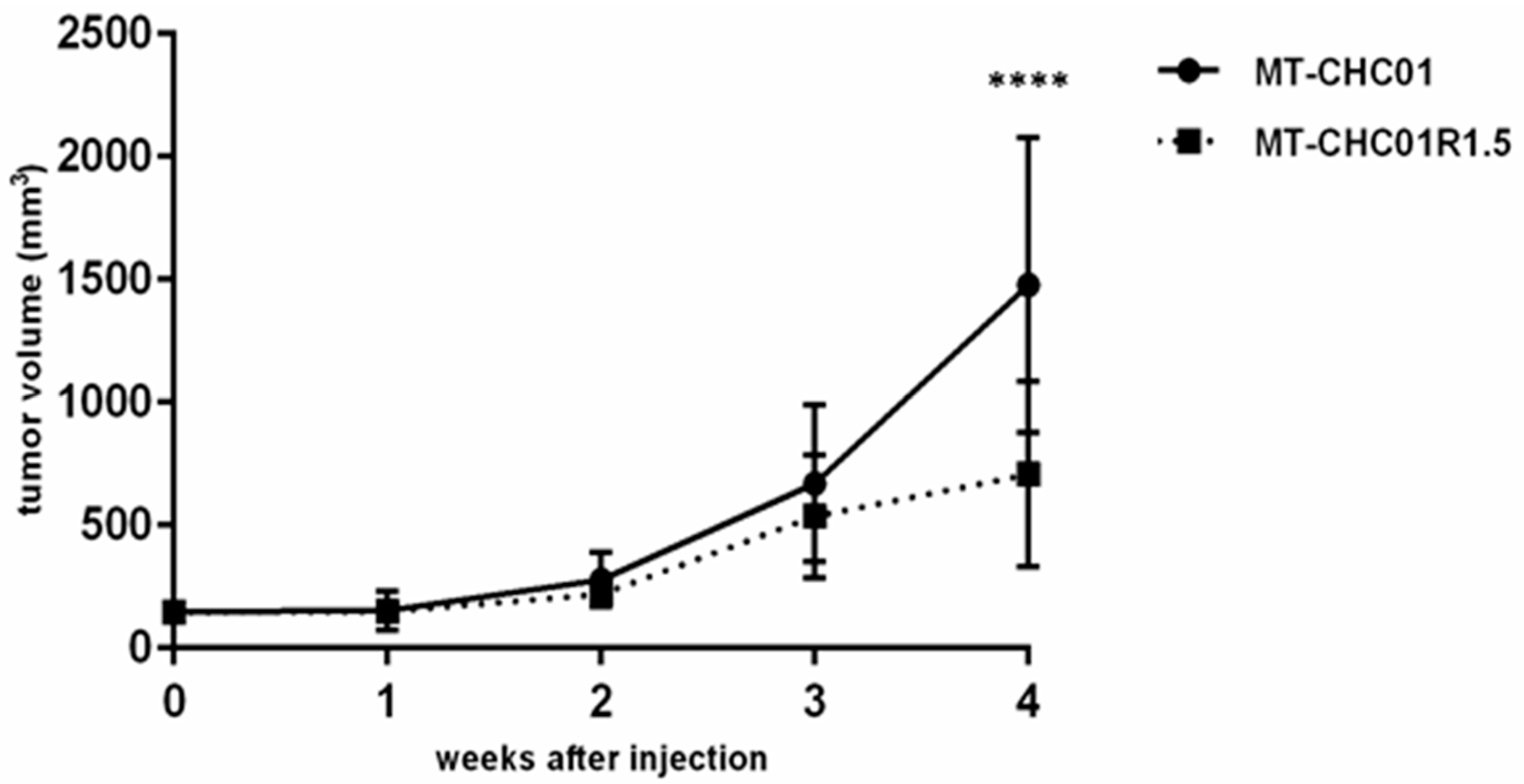
2.1. Biological Characterization of a New Established Gemcitabine Resistant Cholangiocarcinoma Cell Line
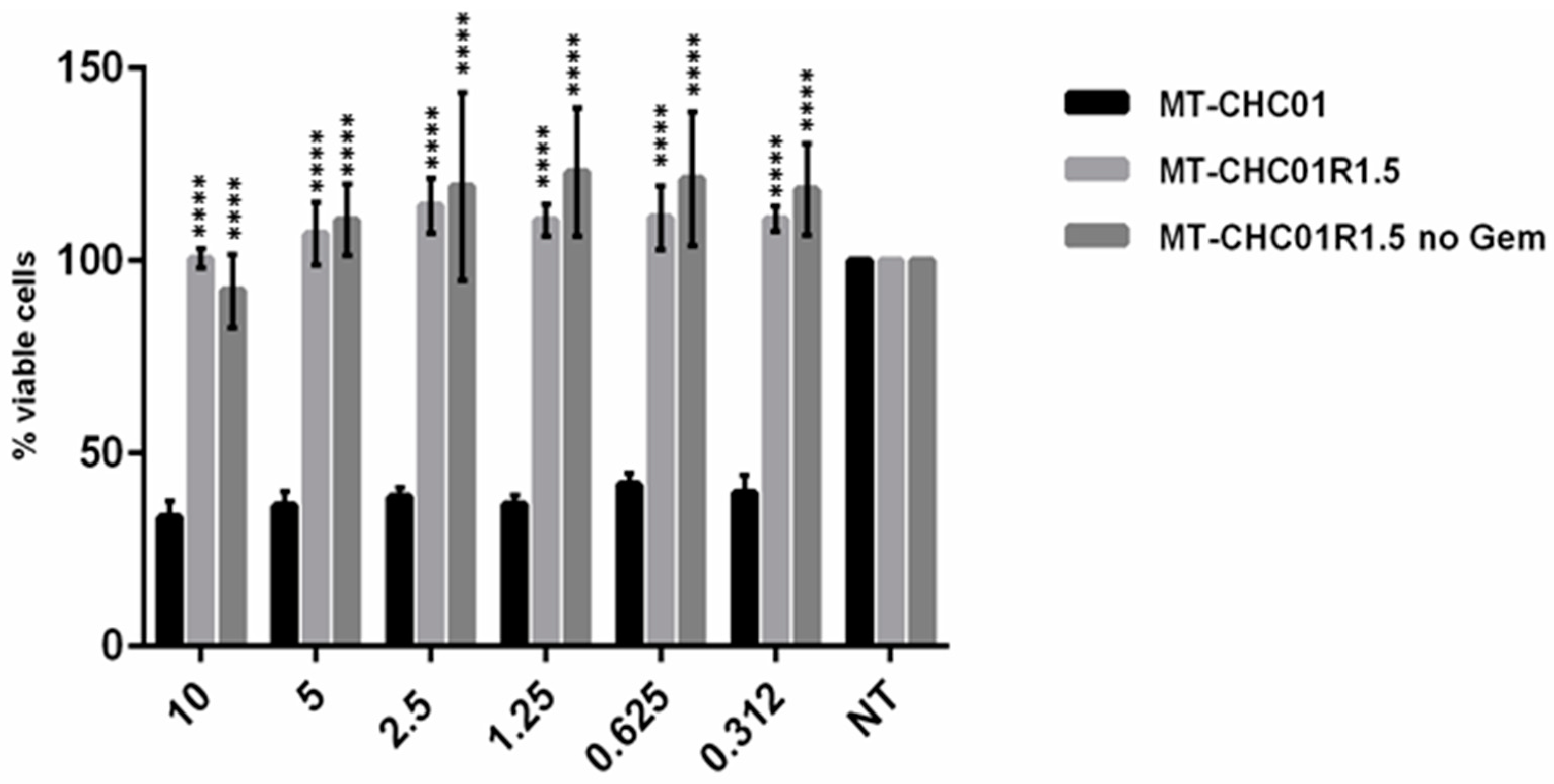
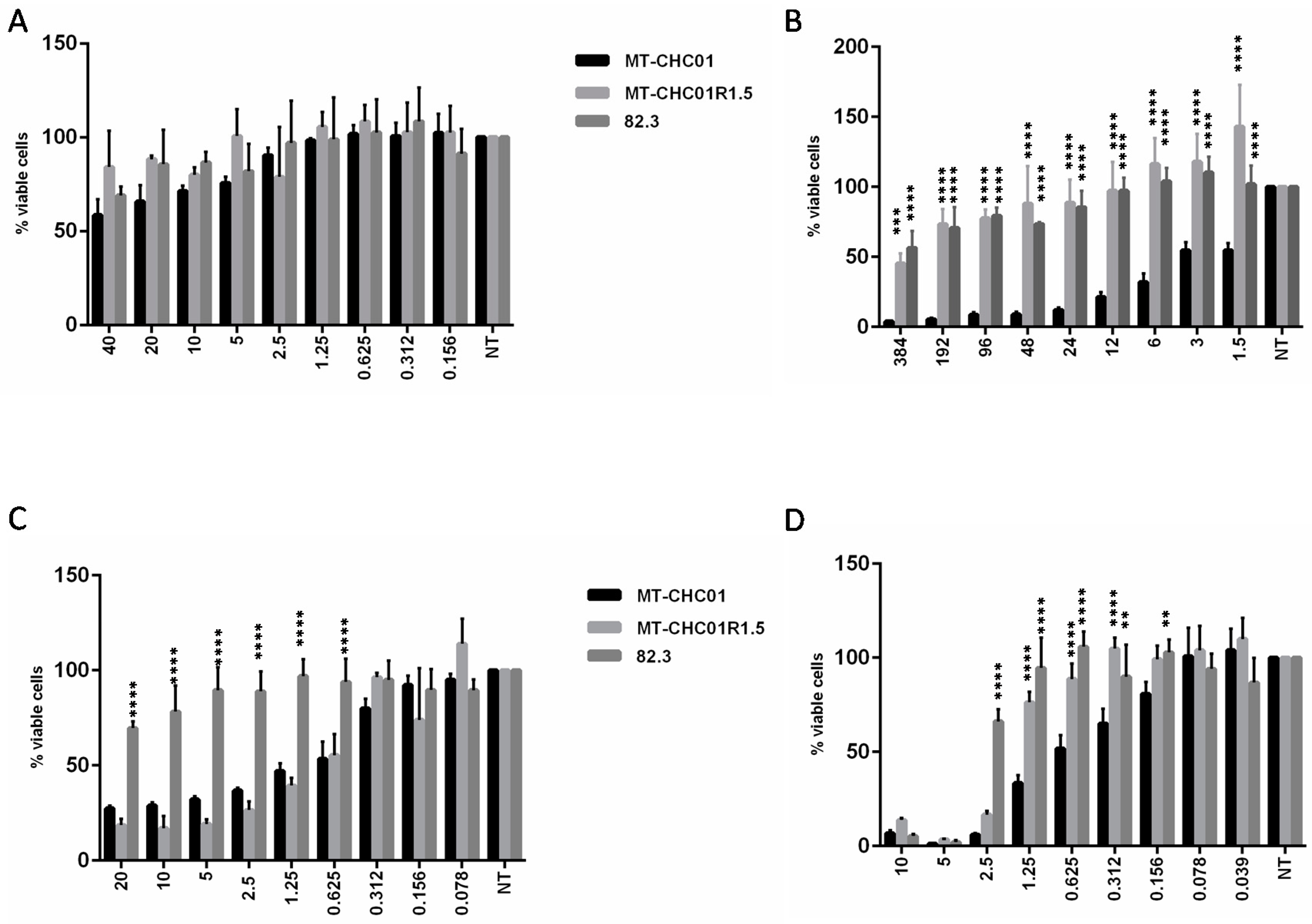
2.2. Gemcitabine Resistant Cells Acquired Cross-Resistance to Other Chemotherapeutic Agents
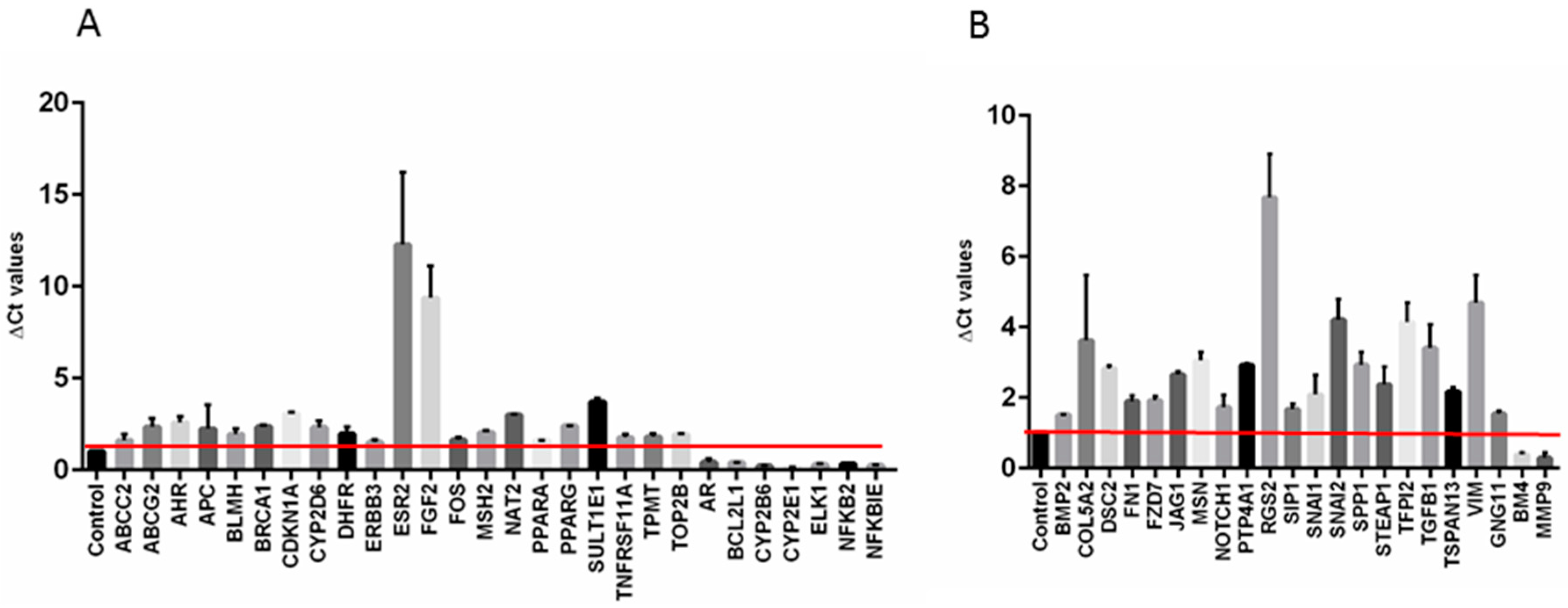
2.3. Gemcitabine Resistant Cells Exhibited Specific Expression Profiles with Important Effects on Biological Processes and Molecular Pathways
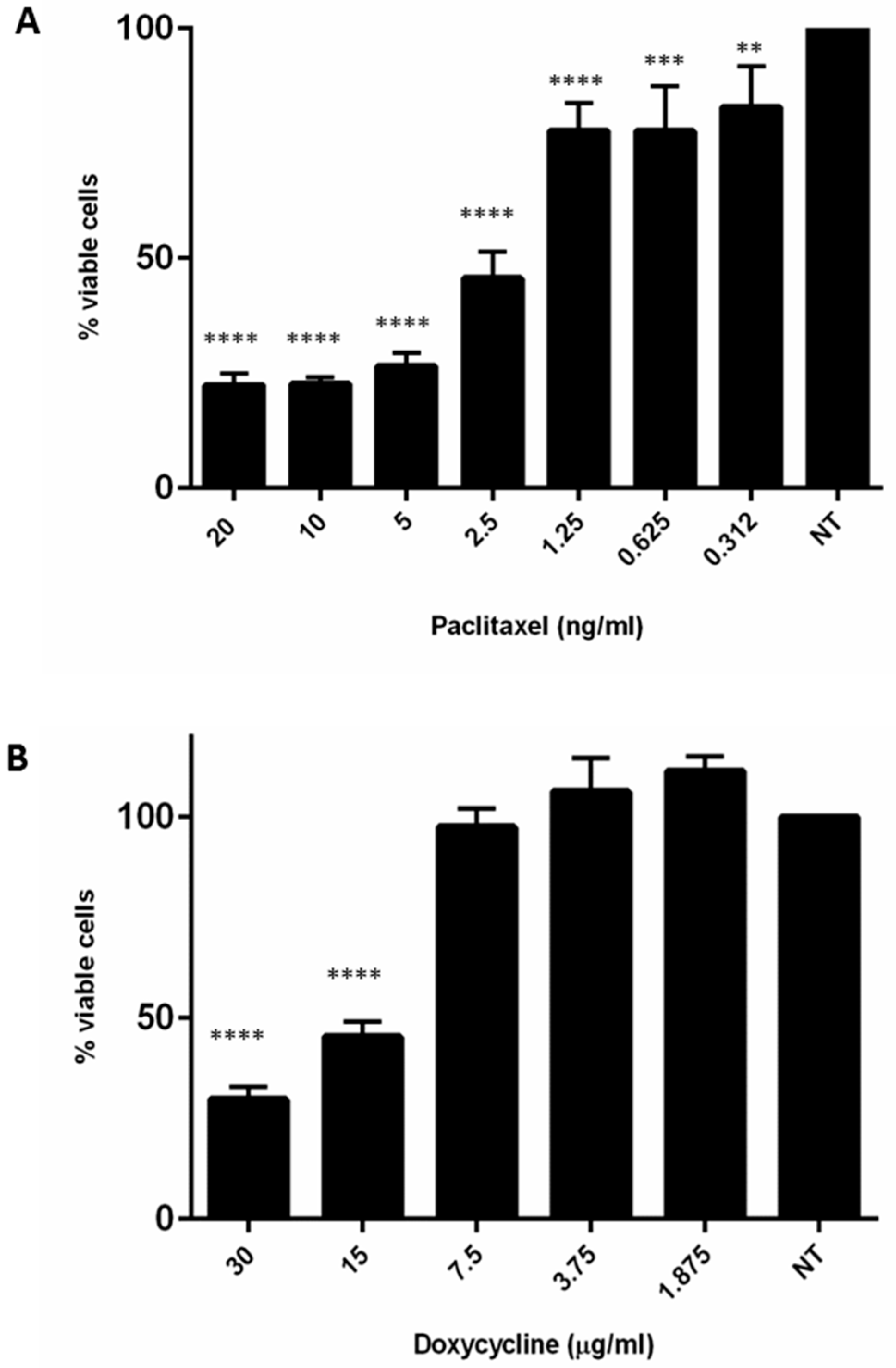
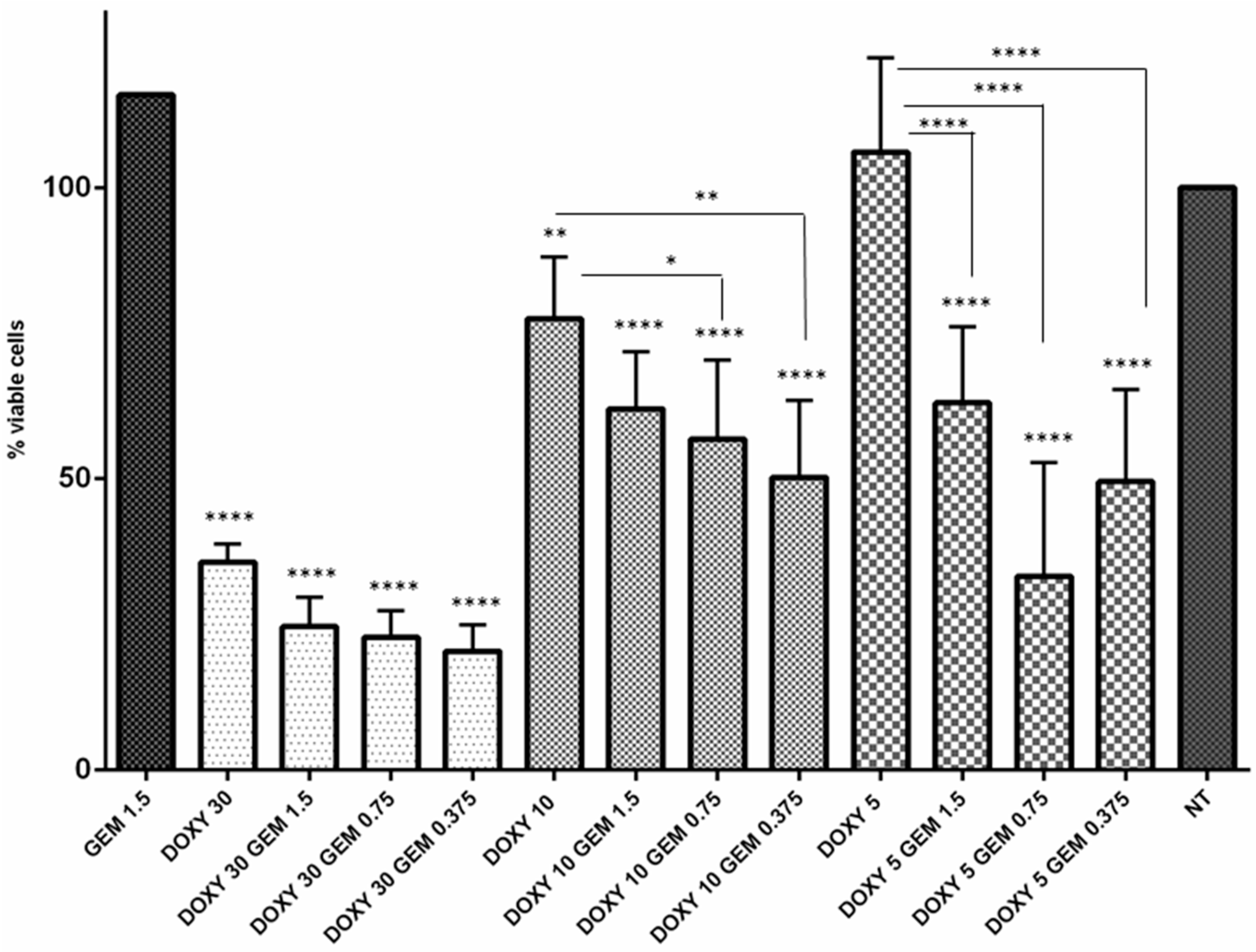
2.4. Doxycycline Restored Gemcitabine Sensitivity in Resistant MT-CHC01R1.5 Cells
2.5. Gemcitabine Resistant Cells Displayed a Modulation of Drug Resistance and Epithelial to Mesenchymal Transitions Related Genes
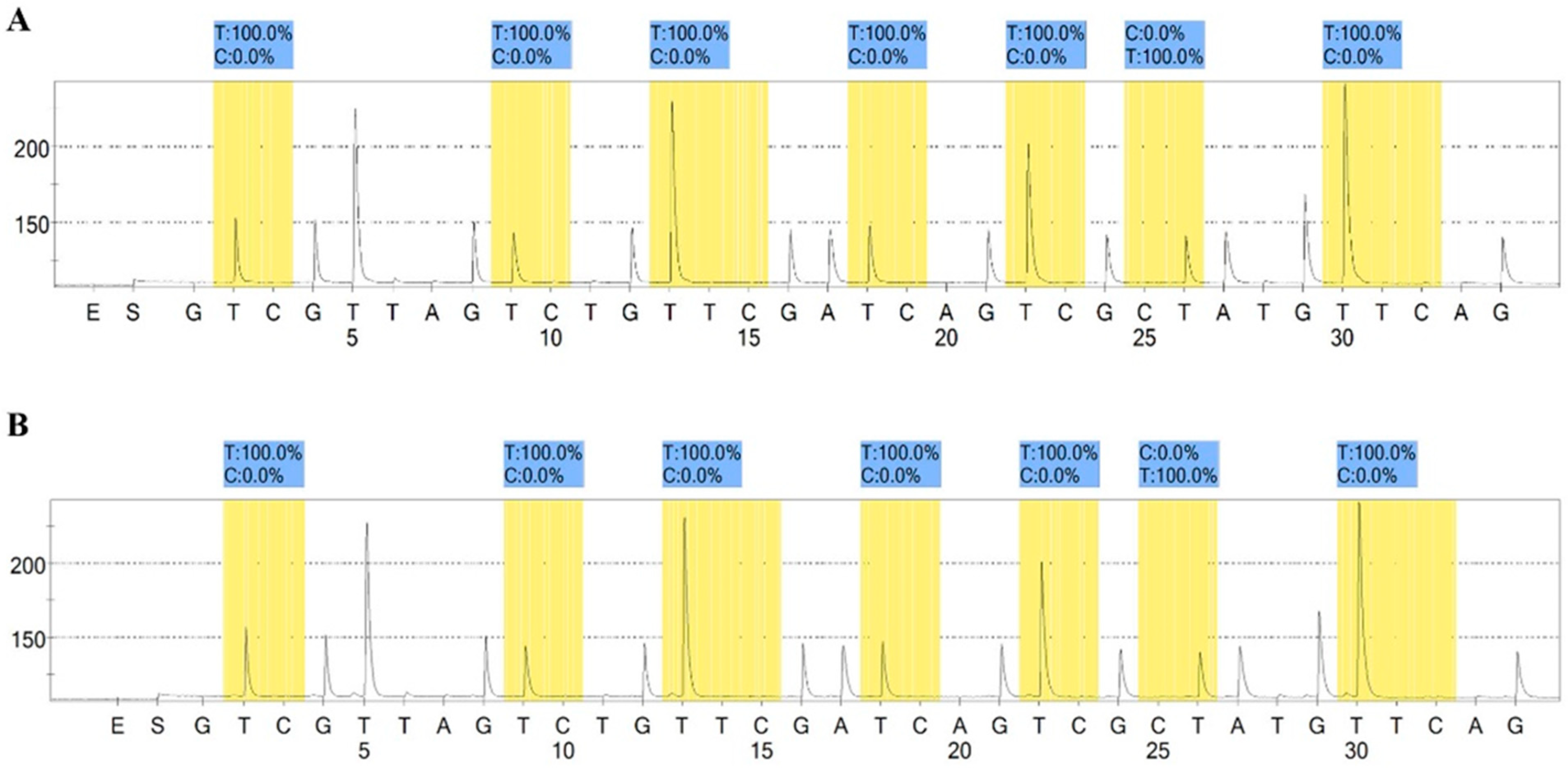
2.6. Methylation Assay
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Establishment of Gemcitabine Resistant Cell Line
4.3. Gemcitabine Resistant ICC Primary Cell Culture
4.4. Flow Cytometry Analysis
4.5. Cell Growth Curve
4.6. Colony Formation Assay
4.7. Cell Growth Assay
4.8. In Vitro Migration and Invasion Assays
4.9. In Vivo Tumor Growth Assay in NOD/SCID Mice
4.10. Gene Expression Analysis (GEP)
4.11. RT2 Profiler PCR Array
4.12. qRT-PCR Validation Assay
4.13. MGMT Promoter Methylation Analyses
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef]
- Valle, J.W.; Furuse, J.; Jitlal, M.; Beare, S.; Mizuno, N.; Wasan, H.; Bridgewater, J.; Okusaka, T. Cisplatin and gemcitabine for advanced biliary tract cancer: A meta-analysis of two randomised trials. Ann. Oncol. 2014, 25, 391–398. [Google Scholar] [CrossRef]
- Eckel, F.; Schmid, R.M. Chemotherapy in advanced biliary tract carcinoma: A pooled analysis of clinical trials. Br. J. Cancer 2007, 96, 896–902. [Google Scholar] [CrossRef]
- Yang, R.; Wang, B.; Chen, Y.J.; Li, H.B.; Hu, J.B.; Zou, S.Q. Efficacy of gemcitabine plus platinum agents for biliary tract cancers: A meta-analysis. Anticancer Drugs 2013, 24, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Taha, R.; Omri, H.E. Current Opinions on Chemoresistance: An Overview. Bioinformation 2018, 14, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Chubb, S.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991, 51, 6110–6117. [Google Scholar]
- Adamska, A.; Elaskalani, O.; Emmanouilidi, A.; Kim, M.; Abdol Razak, N.B.; Metharom, P.; Falasca, M. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv. Biol. Regul. 2018, 68, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Fraveto, A.; Cardinale, V.; Bragazzi, M.C.; Giuliante, F.; De Rose, A.M.; Grazi, G.L.; Napoletano, C.; Semeraro, R.; Lustri, A.M.; Costantini, D.; et al. Sensitivity of Human Intrahepatic Cholangiocarcinoma Subtypes to Chemotherapeutics and Molecular Targeted Agents: A Study on Primary Cell Cultures. PLoS ONE 2015, 10, e0142124. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Y.; Liu, W.; Zhao, G.; Lee, S.; Balogh, A.; Zou, Y.; Guo, Y.; Zhang, Z.; Gu, W.; et al. Doxycycline inducible Krüppel-like factor 4 lentiviral vector mediates mesenchymal to epithelial transition in ovarian cancer cells. PLoS ONE 2014, 9, e105331. [Google Scholar] [CrossRef]
- Maroni, L.; Pierantonelli, I.; Banales, J.M.; Benedetti, A.; Marzioni, M. The significance of genetics for cholangiocarcinoma development. Ann. Transl. Med. 2013, 1, 28. [Google Scholar] [CrossRef]
- Yang, B.; House, M.G.; Guo, M.; Herman, J.G.; Clark, D.P. Promoter methylation profiles of tumor suppressor genes in intrahepatic and extrahepatic cholangiocarcinoma. Mod. Pathol. 2005, 18, 412–420. [Google Scholar] [CrossRef]
- Wattanawongdon, W.; Hahnvajanawong, C.; Namwat, N.; Kanchanawat, S.; Boonmars, T.; Jearanaikoon, P.; Leelayuwat, C.; Techasen, A.; Seubwai, W. Establishment and characterization of gemcitabine-resistant human cholangiocarcinoma cell lines with multidrug resistance and enhanced invasiveness. Int. J. Oncol. 2015, 47, 398–410. [Google Scholar] [CrossRef]
- Ioannou, N.; Seddon, A.M.; Dalgleish, A.; Mackintosh, D.; Solca, F.; Modjtahedi, H. Acquired resistance of pancreatic cancer cells to treatment with gemcitabine and HER-inhibitors is accompanied by increased sensitivity to STAT3 inhibition. Int. J. Oncol. 2016, 48, 908–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhang, W.; Fu, M.; Yang, A.; Huang, H.; Xie, J. Establishment of human pancreatic cancer gemcitabine-resistant cell line with ribonucleotide reductase overexpression. Oncol. Rep. 2015, 33, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Hu, S.S.; Dong, Q.; Cai, J.X.; Zhang, W.P.; Sun, J.Y.; Wang, T.T.; Xie, J.; He, H.R.; Xing, J.F.; et al. Establishment of paclitaxel-resistant breast cancer cell line and nude mice models, and underlying multidrug resistance mechanisms in vitro and in vivo. Asian Pac. J. Cancer Prev. 2013, 14, 6135–6140. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Goessl, E.; Jin, G.; Collie-Duguid, E.S.; Cassidy, J.; Wang, W.; O’Brien, V. Cell cycle perturbation and acquired 5-fluorouracil chemoresistance. Anticancer Res. 2008, 28, 9–14. [Google Scholar] [PubMed]
- Bizama, C.; García, P.; Espinoza, J.A.; Weber, H.; Leal, P.; Nervi, B.; Roa, J.C. Targeting specific molecular pathways holds promise for advanced gallbladder cancer therapy. Cancer Treat. Rev. 2015, 41, 222–234. [Google Scholar] [CrossRef]
- Chiorean, E.G.; Ramasubbaiah, R.; Yu, M.; Picus, J.; Bufill, J.A.; Tong, Y.; Coleman, N.; Johnston, E.L.; Currie, C.; Loehrer, P.J. Phase II trial of erlotinib and docetaxel in advanced and refractory hepatocellular and biliary cancers: Hoosier Oncology Group GI06-101. Oncologist 2012, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Rankin, C.; Siegel, A.B.; Iqbal, S.; Gong, I.Y.; Micetich, K.C.; Kayaleh, O.R.; Lenz, H.J.; Blanke, C.D. S0941: A phase 2 SWOG study of sorafenib and erlotinib in patients with advanced gallbladder carcinoma or cholangiocarcinoma. Br. J. Cancer 2014, 110, 882–887. [Google Scholar] [CrossRef]
- Malka, D.; Cervera, P.; Foulon, S.; Trarbach, T.; de la Fouchardière, C.; Boucher, E.; Fartoux, L.; Faivre, S.; Blanc, J.F.; Viret, F.; et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): A randomised, open-label, non-comparative phase 2 trial. Lancet Oncol. 2014, 15, 819–828. [Google Scholar] [CrossRef]
- Hirose, A.; Tajima, H.; Ohta, T.; Tsukada, T.; Okamoto, K.; Nakanuma, S.; Sakai, S.; Kinoshita, J.; Makino, I.; Furukawa, H.; et al. Low-dose paclitaxel inhibits the induction of epidermal-mesenchymal transition in the human cholangiocarcinoma CCKS-1 cell line. Oncol. Lett. 2013, 6, 915–920. [Google Scholar] [CrossRef] [Green Version]
- Cadamuro, M.; Spagnuolo, G.; Sambado, L.; Indraccolo, S.; Nardo, G.; Rosato, A.; Brivio, S.; Caslini, C.; Stecca, T.; Massani, M.; et al. Low-Dose Paclitaxel Reduces S100A4 Nuclear Import to Inhibit Invasion and Hematogenous Metastasis of Cholangiocarcinoma. Cancer Res. 2016, 76, 4775–4784. [Google Scholar] [CrossRef]
- Wu, W.; Yu, L.H.; Ma, B.; Xu, M.J. The inhibitory effect of doxycycline on cisplatin-sensitive and -resistant epithelial ovarian cancer. PLoS ONE 2014, 9, e89841. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, C.; Liu, X.; He, Y.; Shen, D.; Luo, Q.; Dong, Y.; Dong, H.; Pang, Z. Effects of matrix metalloproteinase inhibitor doxycycline and CD147 antagonist peptide-9 on gallbladder carcinoma cell lines. Tumour Biol. 2017, 39, 1010428317718192. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, P.; Justice, R.; Johnson, J.; Sridhara, R.; Keegan, P.; Pazdur, R. US Food and Drug Administration approval overview in metastatic breast cancer. J. Clin. Oncol. 2012, 30, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Satouchi, M.; Okamoto, I.; Sakai, H.; Yamamoto, N.; Ichinose, Y.; Ohmatsu, H.; Nogami, N.; Takeda, K.; Mitsudomi, T.; Kasahara, K.; et al. Efficacy and safety of weekly nab-paclitaxel plus carboplatin in patients with advanced non-small cell lung cancer. Lung Cancer 2013, 81, 97–101. [Google Scholar] [CrossRef]
- Unseld, M.; Scheithauer, W.; Weigl, R.; Kornek, G.; Stranzl, N.; Bianconi, D.; Brunauer, G.; Steger, G.; Zielinski, C.C.; Prager, G.W. Nab-paclitaxel as alternative treatment regimen in advanced cholangiocellular carcinoma. J. Gastrointest. Oncol. 2016, 7, 588–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, M.C.; Hong, S.M.; Augustine, M.M.; Goggins, M.G.; Wolfgang, C.L.; Hirose, K.; Schulick, R.D.; Choti, M.A.; Anders, R.A.; Pawlik, T.M. Hilar cholangiocarcinoma: Tumor depth as a predictor of outcome. Arch. Surg. 2011, 146, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, X.; Wang, H.; Zhou, Q.; Liang, Y.; Sui, A.; Yao, R.; Zhao, B.; Sun, M. Lentiviral vector-mediated doxycycline-inducible USP39 shRNA or cDNA expression in triple-negative breast cancer cells. Oncol. Rep. 2015, 33, 2477–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, K.; Fujioka, S.; Iida, T.; Furukawa, K.; Fujita, T.; Yamada, H.; Chiao, P.J.; Yanaga, K. Doxycycline induces apoptosis in PANC-1 pancreatic cancer cells. Anticancer Res. 2009, 29, 3995–4003. [Google Scholar] [PubMed]
- De Francesco, E.M.; Maggiolini, M.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Targeting hypoxic cancer stem cells (CSCs) with Doxycycline: Implications for optimizing anti-angiogenic therapy. Oncotarget 2017, 8, 56126–56142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujioka, S.; Son, K.; Onda, S.; Schmidt, C.; Scrabas, G.M.; Okamoto, T.; Fujita, T.; Chiao, P.J.; Yanaga, K. Desensitization of NFκB for overcoming chemoresistance of pancreatic cancer cells to TNF-α or paclitaxel. Anticancer Res. 2012, 32, 4813–4821. [Google Scholar] [PubMed]
- Deng, T.; Pan, H.; Han, R.; Huang, D.; Li, H.; Zhou, L.; Wang, X.; Bai, M.; Li, X.; Liu, R.; et al. Gemcitabine sensitivity factors, hENT1 and RRM1 as potential prognostic biomarker for advanced biliary tract cancer. Int. J. Clin. Exp. Med. 2014, 7, 5041–5049. [Google Scholar]
- Tsai, M.S.; Kuo, Y.H.; Chiu, Y.F.; Su, Y.C.; Lin, Y.W. Down-regulation of Rad51 expression overcomes drug resistance to gemcitabine in human non-small-cell lung cancer cells. J. Pharmacol. Exp. Ther. 2010, 335, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef]
- Ueno, H.; Kiyosawa, K.; Kaniwa, N. Pharmacogenomics of gemcitabine: Can genetic studies lead to tailor-made therapy? Br. J. Cancer 2007, 97, 145–151. [Google Scholar] [CrossRef]
- Colavito, D.; Cartei, G.; Dal Bianco, M.; Stecca, A.; Zustovich, F.; Dalle Carbonare, M.; Ragazzi, E.; Farina, M.; Colombrino, E.; Leon, A. Thymidylate synthetase allelic imbalance in clear cell renal carcinoma. Cancer Chemother. Pharmacol. 2009, 64, 1195–1200. [Google Scholar] [CrossRef]
- Mehrmohamadi, M.; Jeong, S.H.; Locasale, J.W. Molecular features that predict the response to antimetabolite chemotherapies. Cancer Metab. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntziachristos, P.; Lim, J.S.; Sage, J.; Aifantis, I. From fly wings to targeted cancer therapies: A centennial for notch signaling. Cancer Cell 2014, 25, 318–334. [Google Scholar] [CrossRef]
- Kim, H.B.; Cho, W.J.; Choi, N.G.; Kim, S.S.; Park, J.H.; Lee, H.J.; Park, S.G. Clinical implications of APEX1 and Jagged1 as chemoresistance factors in biliary tract cancer. Ann. Surg. Treat. Res. 2017, 92, 15–22. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D.; Grompe, M. The Fanconi anaemia/BRCA pathway. Nat. Rev. Cancer 2003, 3, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, S.; Kobayashi, S.; Nagano, H.; Tomokuni, A.; Tomimaru, Y.; Asaoka, T.; Hama, N.; Wada, H.; Kawamoto, K.; Marubashi, S.; et al. BRCA/Fanconi anemia pathway implicates chemoresistance to gemcitabine in biliary tract cancer. Cancer Sci. 2015, 106, 584–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, W.H.; Zeng, P.; Chen, X.; Lu, Y.J.; Li, A.; Wu, J.B. Promotive effects of bone morphogenetic protein 2 on angiogenesis in hepatocarcinoma via multiple signal pathways. Sci. Rep. 2016, 6, 37499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, H.; Luo, H.; Wang, H.; Chen, X.; Li, P.; Bai, Y.; Zhang, F.; Cheng, R.; Chen, S.; Wang, Y.; et al. Silencing BMP-2 expression inhibits A549 and H460 cell proliferation and migration. Diagn. Pathol. 2014, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Hanyu, A.; Kanda, H.; Ishikawa, Y.; Akiyama, F.; Iwase, T.; Ogata, E.; Ehata, S.; Miyazono, K.; Imamura, T. Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through Smad pathway. Oncogene 2008, 27, 6322–6333. [Google Scholar] [CrossRef] [Green Version]
- Vishnubalaji, R.; Yue, S.; Alfayez, M.; Kassem, M.; Liu, F.F.; Aldahmash, A.; Alajez, N.M. Bone morphogenetic protein 2 (BMP2) induces growth suppression and enhances chemosensitivity of human colon cancer cells. Cancer Cell Int. 2016, 16, 77. [Google Scholar] [CrossRef]
- Choi, Y.J.; Ingram, P.N.; Yang, K.; Coffman, L.; Iyengar, M.; Bai, S.; Thomas, D.G.; Yoon, E.; Buckanovich, R.J. Identifying an ovarian cancer cell hierarchy regulated by bone morphogenetic protein 2. Proc. Natl. Acad. Sci. USA 2015, 112, E6882–E6888. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.J.; Kuo, Y.L.; Chen, C.C.; Chen, M.J.; Cheng, Y.M. MMP1 expression is activated by Slug and enhances multi-drug resistance (MDR) in breast cancer. PLoS ONE 2017, 12, e0174487. [Google Scholar] [CrossRef]
- Yang, J.M.; Xu, Z.; Wu, H.; Zhu, H.; Wu, X.; Hait, W.N. Overexpression of extracellular matrix metalloproteinase inducer in multidrug resistant cancer cells. Mol. Cancer Res. 2003, 1, 420–427. [Google Scholar]
- Zhang, J.; Yin, D.; Li, H. hMSH2 expression is associated with paclitaxel resistance in ovarian carcinoma, and inhibition of hMSH2 expression in vitro restores paclitaxel sensitivity. Oncol. Rep. 2014, 32, 2199–2206. [Google Scholar] [CrossRef]
- Chauhan, P.S.; Bhushan, B.; Singh, L.C.; Mishra, A.K.; Saluja, S.; Mittal, V.; Gupta, D.K.; Kapur, S. Expression of genes related to multiple drug resistance and apoptosis in acute leukemia: Response to induction chemotherapy. Exp. Mol. Pathol. 2012, 92, 44–49. [Google Scholar] [CrossRef]
- Sherman-Baust, C.A.; Becker, K.G.; Wood Iii, W.H.; Zhang, Y.; Morin, P.J. Gene expression and pathway analysis of ovarian cancer cells selected for resistance to cisplatin, paclitaxel, or doxorubicin. J. Ovarian Res. 2011, 4, 21. [Google Scholar] [CrossRef]
- Cavalloni, G.; Peraldo-Neia, C.; Varamo, C.; Casorzo, L.; Dell’Aglio, C.; Bernabei, P.; Chiorino, G.; Aglietta, M.; Leone, F. Establishment and characterization of a human intrahepatic cholangiocarcinoma cell line derived from an Italian patient. Tumour Biol. 2015, 37, 4041–4052. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Networks | p-Value | Genes Involved |
|---|---|---|
| Cell cycle_S phase | 1.2 × 10−24 | MCM4, Histone H1.5, Histone H4, RFC5, MCM3, CHTF18, Rad51, Cdt1, MCM2, PCNA, FEN1, ORC1L, RFC2, MCM7, ChAF1 subunit A, MCM6, Histone H1, POLD cat (p125), POLA2, DNA ligase I, PRIM1, MCM5, MCM8, E2F1, CDC45L, MCM4/6/7 complex, ChAF1 subunit B |
| Cell cycle_Core | 6.9 × 10−13 | E2F2, MCM4, MCM3, Cdt1, MCM2, FEN1, Kinase MYT1, ORC1L, MCM7, MCM6, DNA ligase I, MCM5, MCM8, E2F1, CDC45L, MCM4/6/7 complex |
| DNA damage_DBS repair | 2.7 × 10−7 | Histone H4, XRCC2, Rad51, PCNA, FEN1, ChAF1 subunit A, RAD54L, FANCD2, FANCA, DNA ligase I, ChAF1 subunit B |
| DNA damage_MMR repair | 8.8 × 10−4 | RFC5, EXO1, PCNA, RFC2, POLD cat (p125) |
| DNA damage_Checkpoint | 9.8 × 10−4 | RFC5, CIA/ASF1, PCNA, Kinase MYT1, RFC2, FANCD2, E2F1 |
| Cell cycle_G1-S | 0.001 | Rad51, TYSY, PCNA, b-Myb, UHRF1, Ceb1, FANCD2, E2F1 |
| DNA damage_BER-NER repair | 0.002 | RFC5, PCNA, FEN1, RFC2, POLD cat (p125), DNA ligase I |
| Apoptosis_Apoptotic nucleus | 0.004 | Histone H1.5, Rad51, c-Myb, b-Myb, Histone H1, FANCD2, E2F1 |
| Reproduction_Spermatogenesis, motility and copulation | 0.008 | Histone H1.1, PKC-beta, Histone H2, Histone H2A, PKC, DDC, Histone H1, FANCA |
| Cytoskeleton_Regulation of cytoskeleton rearrangement | 0.008 | Tubulin beta, Vimentin, Talin, PKC, Tubulin beta 1, Tubulin beta 2, Band 4.1-like protein 2 |
| Maps | p-Value | Genes Involved |
|---|---|---|
| Cell cycle_Start of DNA replication in early S phase | 2.68 × 10−14 | MCM4, MCM3, Cdt1, MCM2, ORC1L, Histone H1, MCM5, E2F1, CDC45L, MCM4/6/7 complex |
| Cell cycle_Transition and termination of DNA replication | 1.86 × 10−6 | MCM2, PCNA, FEN1, POLD cat (p125), DNA ligase I |
| NETosis in SLE | 3.7 × 10−6 | Histone H4, Histone H2, Histone H2A, PKC, Histone H1 |
| dCTP/dUTP metabolism | 2.29 × 10−5 | Ribonucleotide reductase, RRM2, Small RR subunit, RRM1, POLD cat (p125), POLA2 |
| dATP/dITP metabolism | 4.8 × 10−5 | Ribonucleotide reductase, RRM2, Small RR subunit, RRM1, POLD cat (p125), POLA2 |
| Cell cycle_Sister chromatid cohesion | 6.3 × 10−4 | CHTF18, PCNA, Histone H1 |
| Immune response_IFN-alpha/beta signaling via PI3K and NF-kB pathways | 6.8 × 10−4 | DHFR, PCNA, b-Myb, E2F1, ISG15 |
| Cytoskeleton remodeling_Neurofilaments | 9.3 × 10−4 | Tubulin beta, Vimentin, NEFL |
| Transcription_Effect of Folic acid on genome stability | 0.001 | DHFR, HCP1, TYSY, ADH1 |
| Immune response_IL-4-induced regulators of cell growth, survival, differentiation and metabolism | 0.001 | MCM4, MCM6, MMP-13, MCM5 |
| Folic acid metabolism | 0.001 | C1TC, DHFR, HCP1, SLC19A1 |
| DNA damage_Role of Brca1 and Brca2 in DNA repair | 0.001 | Rad51, PCNA, FANCD2 |
| Development_Transcription regulation of granulocyte development | 0.001 | c-Myb, PKC, E2F1 |
| Transcription_Negative regulation of HIF1A function | 0.002 | MCM3, MCM2, MCM7, MCM5 |
| Inhibition of neutrophil migration by proresolving lipid mediators in COPD | 0.002 | TLN2, Talin, PKC, cPKC |
| Signal transduction_HTR2A signaling outside the nervous system | 0.002 | PKC-beta, PKC, cPKC (conventional), E2F1 |
| DNA damage_Nucleotide excision repair | 0.003 | PCNA, POLD cat (p125), DNA ligase I |
| Cell adhesion_Cell-matrix glycoconjugates | 0.003 | CCL5, MMP-1, ECM1 |
| Development_Gastrin in differentiation of the gastric mucosa | 0.003 | PKC-beta, PKC, cPKC |
| Complement pathway disruption in thrombotic microangiopathy | 0.003 | CCL5, PKC, cPKC |
| Immune response_IL-1 signaling pathway | 0.003 | CCL5, SPHK1, MMP-1, MMP-13 |
| Oxidative stress_Activation of NOX1, NOX5, DUOX1 and DUOX2 NADPH Oxidases | 0.004 | PKC-beta, PKC, cPKC |
| Development_VEGF signaling and activation | 0.004 | PKC-beta, SPHK1, PKC |
| Glomerular injury in Lupus Nephritis | 0.005 | CCL5, PKC-beta1, MMP-1, PKC-beta2 |
| Apoptosis and survival_DNA-damage-induced apoptosis | 0.005 | FANCD2, E2F1 |
| Transcription_Assembly of RNA Polymerase II preinitiation complex on TATA-less promoters | 0.008 | DHFR, PCNA |
| Immune response_IL-16 signaling pathway | 0.009 | CCL5, PKC, cPKC |
| DNA damage_Mismatch repair | 0.009 | EXO1, PCNA |
| Networks | p-Value | Genes Involved |
|---|---|---|
| Reproduction_Gonadotropin regulation | 0.003 | ATF-3, EGR1, HB-EGF, GABA-A receptor epsilon subunit, Adenylate cyclase, FosB, AKR1C3, c-Fos |
| Proteolysis_Connective tissue degradation | 0.004 | Matrilysin (MMP-7), Trypsin II, Trypsin, ADAM8, SERPINA3 (ACT), Protein C inhibitor |
| Reproduction_FSH-beta signaling pathway | 0.004 | EGR1, TGM2, Adenylate cyclase, VEGF-A, ActRIIB, c-Fos, IBP |
| Signal transduction_ESR1-membrane pathway | 0.005 | HB-EGF, Adenylate cyclase, Adenylate cyclase type IV, c-Fos, Caveolin-1 |
| Development_Regulation of angiogenesis | 0.007 | Ephrin-A, Ephrin-A1, IL-15, Angiotensin II, HB-EGF, Angiogenin, VEGF-A, Angiotensin III |
| Signal transduction_WNT signaling | 0.007 | Matrilysin (MMP-7), HES1, WISP2, HB-EGF, Adenylate cyclase, VEGF-A, c-Fos |
| Maps | p-Value | Genes Involved |
|---|---|---|
| Protein folding and maturation_Angiotensin system maturation | 2.223 ×10−8 | Angiotensin (2–10), Angiotensin II, Angiotensin IV, Angiotensin I, Angiotensin (1–7), Angiotensinogen, Angiotensin III, Angiotensin (1–9) |
| Protein folding and maturation_Posttranslational processing of neuroendocrine peptides | 1.017 × 10−7 | NT, LargeNT, Trypsin, NN, LargeNN, NT/NN |
| Transcription_HIF-1 targets | 4.662 × 10−5 | TGM2, MCT4, IBP1, Stanniocalcin 2, VEGF-A, Cyclin G2, REDD1 |
| Signal transduction_HTR2A signaling outside the nervous system | 9.750 × 10−5 | EGR1, HB-EGF, Adenylate cyclase, PLD1, HB-EGF (mature), Caveolin-1 |
| Signal transduction_mTORC1 downstream signaling | 3.624 × 10−4 | eIF4A, VEGF-A, PDCD4, eIF4B, ULK1 |
| Immune response_CD16 signaling in NK cells | 6.423 × 10−4 | PLA2, PLD1, SHIP, c-Fos, VAV-1 |
| Reproduction_Gonadotropin-releasing hormone (GnRH) signaling | 7.808 × 10−4 | ATF-3, EGR1, Adenylate cyclase, FosB, c-Fos |
| L-Tryptophan metabolism (part 1) | 0.001 | SLC43A1, ASCT2 (SLC1A5), SLC7A8, SLC38A2, SLC6A14 |
| Development_Leptin signaling via JAK/STAT and MAPK cascades | 0.001 | EGR1, VEGF-A, c-Fos |
| Apoptosis and survival_Apoptotic Activin A signaling | 0.001 | SHIP, ActRIIB, c-Fos |
| Putative pathways of hormone action in neurofibromatosis type 1 | 0.001 | EGR1, EGR2 (Krox20), FosB |
| Membrane-bound ESR1: interaction with G-proteins signaling | 0.002 | HB-EGF, Adenylate cyclase, c-Fos, Caveolin-1 |
| Development_Non-genomic action of Retinoic acid in cell differentiation | 0.002 | TGM2, VEGF-A, c-Fos, VAV-1 |
| IGF family signaling in colorectal cancer | 0.003 | FosB, VEGF-A, c-Fos, IBP |
| Regulation of lipid metabolism_FXR-dependent negative-feedback regulation of bile acids concentration | 0.003 | FGF19, CYP3A4, CYP2B6 |
| Cytoskeleton remodeling_Fibronectin-binding integrins in cell motility | 0.004 | ITGAV, VAV-1, Caveolin-1 |
| Development_Role of IL-8 in angiogenesis | 0.004 | HB-EGF, VEGF-A, c-Fos, Caveolin-1 |
| Muscle contraction_Regulation of eNOS activity in endothelial cells | 0.004 | KLF2, FosB, VEGF-A, Caveolin-1 |
| Development_Angiotensin activation of ERK | 0.004 | Angiotensin II, HB-EGF, c-Fos |
| Immune response_IL-6-induced acute-phase response in hepatocytes | 0.005 | Angiotensinogen, IBP1, c-Fos |
| Retinol metabolism | 0.005 | CYP3A5, CYP3A4, CYP2B6, CYP3A7 |
| Immune response_IL-6 signaling pathway via JAK/STAT | 0.005 | Rac2, VEGF-A, c-Fos, VAV-1 |
| Transport_ACM3 signaling in lacrimal glands | 0.006 | Mucin 5B, PLD1, Aquaporin 5, Lysozyme |
| Transcription_P53 signaling pathway | 0.007 | HSP27, VEGF-A, c-Fos |
| Role of tumor microenvironment in plexiform neurofibroma formation in neurofibromatosis type 1 | 0.007 | Rac2, VEGF-A, VAV-1 |
| Development_Role of Activin A in cell differentiation and proliferation | 0.007 | p15, Adenylate cyclase, ActRIIB |
| Main pathways of Schwann cells transformation in neurofibromatosis type 1 | 0.008 | Rac2, VEGF-A, c-Fos, VAV-1 |
| Development_Angiotensin signaling via PYK2 | 0.009 | Angiotensin II, c-Fos, VAV-1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varamo, C.; Peraldo-Neia, C.; Ostano, P.; Basiricò, M.; Raggi, C.; Bernabei, P.; Venesio, T.; Berrino, E.; Aglietta, M.; Leone, F.; et al. Establishment and Characterization of a New Intrahepatic Cholangiocarcinoma Cell Line Resistant to Gemcitabine. Cancers 2019, 11, 519. https://doi.org/10.3390/cancers11040519
Varamo C, Peraldo-Neia C, Ostano P, Basiricò M, Raggi C, Bernabei P, Venesio T, Berrino E, Aglietta M, Leone F, et al. Establishment and Characterization of a New Intrahepatic Cholangiocarcinoma Cell Line Resistant to Gemcitabine. Cancers. 2019; 11(4):519. https://doi.org/10.3390/cancers11040519
Chicago/Turabian StyleVaramo, Chiara, Caterina Peraldo-Neia, Paola Ostano, Marco Basiricò, Chiara Raggi, Paola Bernabei, Tiziana Venesio, Enrico Berrino, Massimo Aglietta, Francesco Leone, and et al. 2019. "Establishment and Characterization of a New Intrahepatic Cholangiocarcinoma Cell Line Resistant to Gemcitabine" Cancers 11, no. 4: 519. https://doi.org/10.3390/cancers11040519
APA StyleVaramo, C., Peraldo-Neia, C., Ostano, P., Basiricò, M., Raggi, C., Bernabei, P., Venesio, T., Berrino, E., Aglietta, M., Leone, F., & Cavalloni, G. (2019). Establishment and Characterization of a New Intrahepatic Cholangiocarcinoma Cell Line Resistant to Gemcitabine. Cancers, 11(4), 519. https://doi.org/10.3390/cancers11040519