Cyclin-Dependent Kinase 5 (CDK5)-Mediated Phosphorylation of Upstream Stimulatory Factor 2 (USF2) Contributes to Carcinogenesis

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
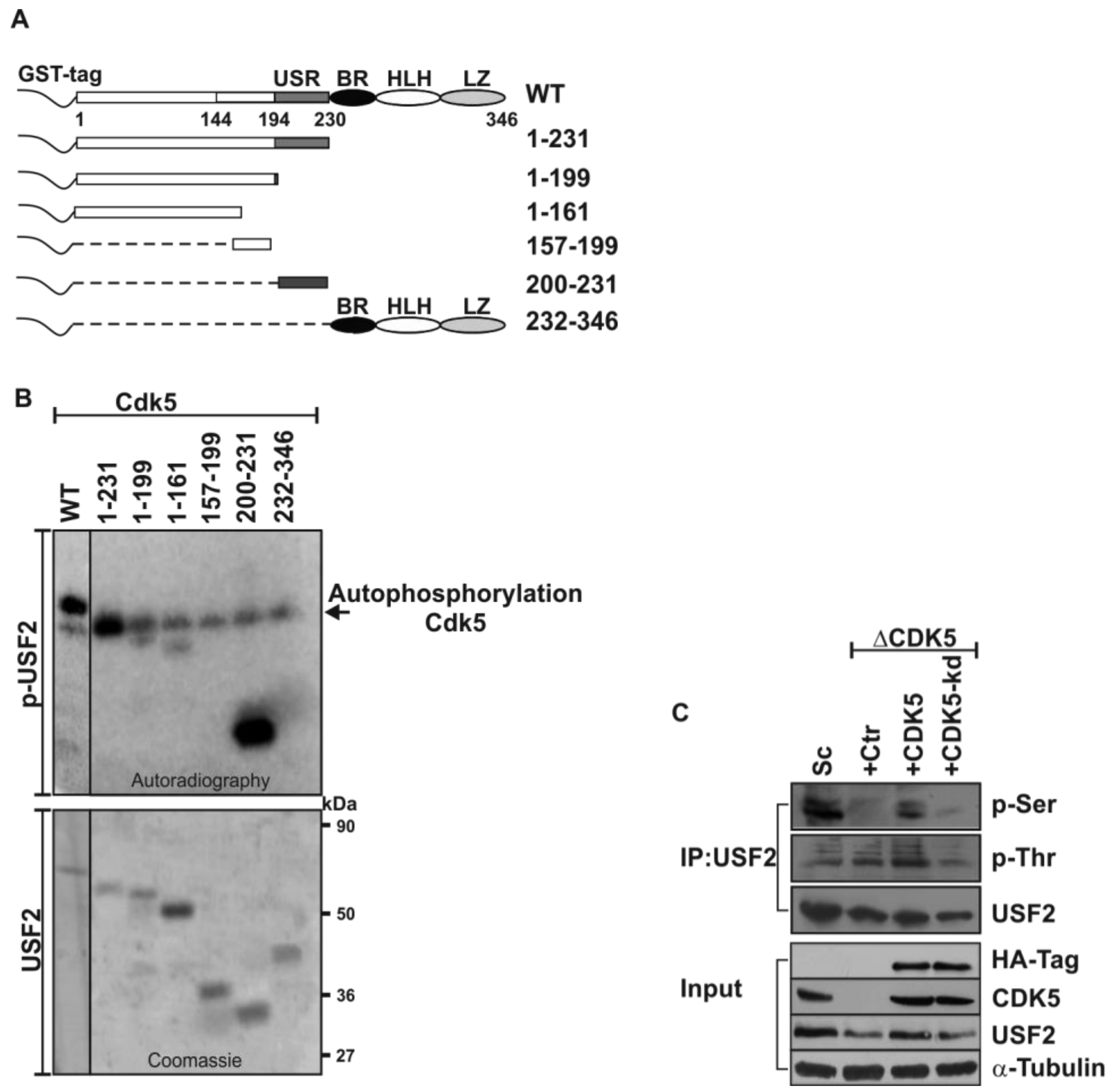
2.1. USF2 is a Phosphorylation Target of CDK5
2.2. Mapping the Exact CDK5 Phosphorylation Sites within USF2
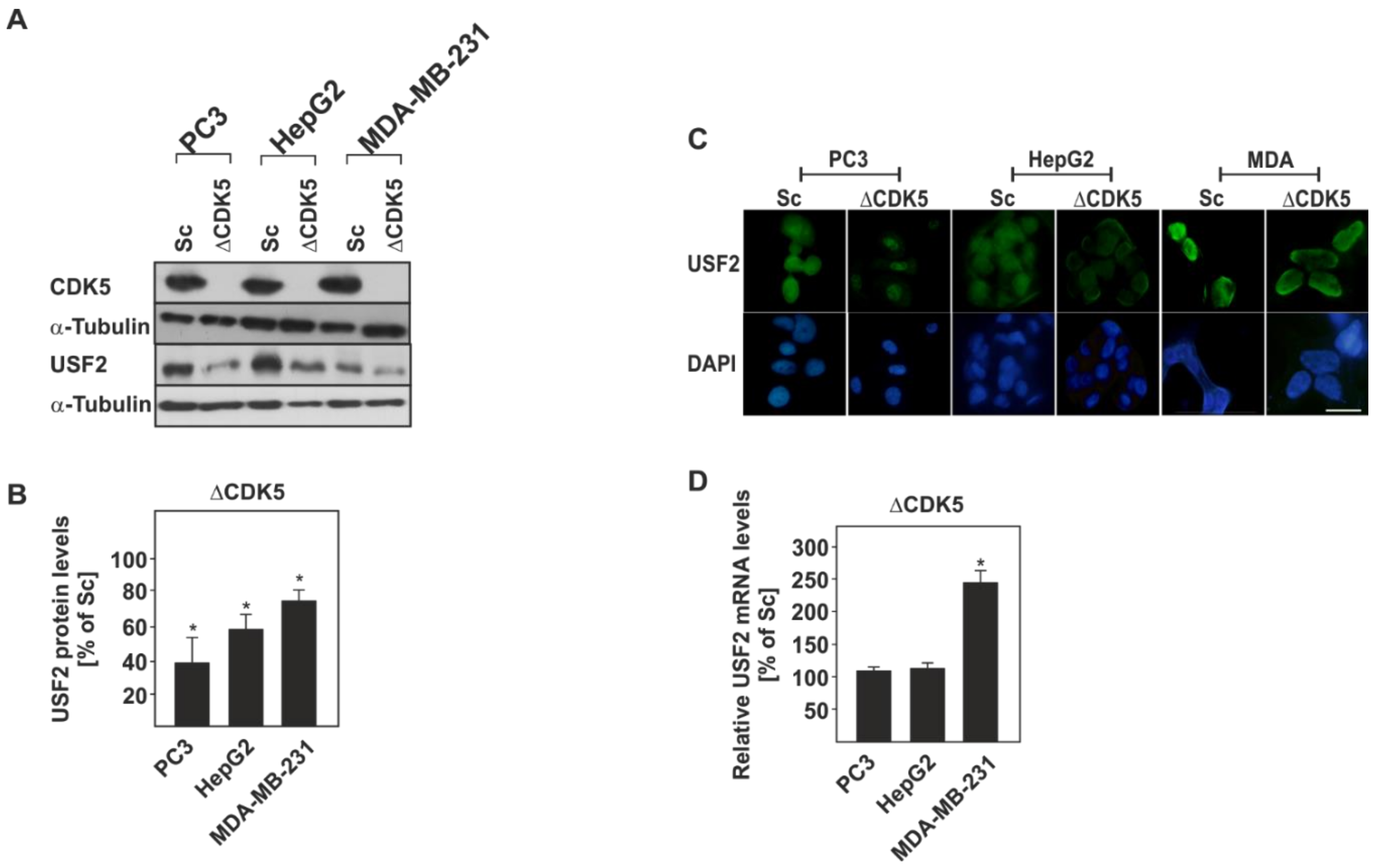
2.3. The Protein Levels of USF2 are Altered in the Absence of CDK5
2.4. Lack of CDK5 Decreases the Half-Life of USF2
2.5. Phosphorylation of USF2 by CDK5 Affects Cell Proliferation and Migration
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Plasmid Constructs
4.3. Cell Culture, Transfection, and Luciferase Assays
4.4. RNA Preparation and Quantitative Real-Time PCR Analyses
4.5. Generation of CDK5 Knockout PC3, HepG2, and MDA-MB-231 Cell Lines by CRISPR-Cas9-Mediated Genome Editing
4.6. Protein Isolation, Immunoprecipitations, Half-Life Studies, Immunofluorescence, and Western Blots
4.7. Expression and Purification of Proteins, Radioactive Kinase Assays
4.8. Overexpression of USF2-S155A/S222A and USF2-S155D/S222D Double Point Mutants in Cells via Lentiviral Infection
4.9. Proliferation and Cell Migration Assay
4.10. Apoptosis Analyses
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Viollet, B.; Lefrancois-Martinez, A.M.; Henrion, A.; Kahn, A.; Raymondjean, M.; Martinez, A. Immunochemical characterization and transacting properties of upstream stimulatory factor isoforms. J. Biol. Chem. 1996, 271, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Sirito, M.; Lin, Q.; Deng, J.M.; Behringer, R.R.; Sawadogo, M. Overlapping roles and asymmetrical cross-regulation of the USF proteins in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3758–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murre, C.; McCaw, P.S.; Vaessin, H.; Caudy, M.; Jan, L.Y.; Jan, Y.N.; Cabrera, C.V.; Buskin, J.N.; Hauschka, S.D.; Lassar, A.B. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell 1989, 58, 537–544. [Google Scholar] [CrossRef]
- Lin, Q.; Luo, X.; Sawadogo, M. Archaic structure of the gene encoding transcription factor USF. J. Biol. Chem. 1994, 269, 23894–23903. [Google Scholar]
- Henrion, A.A.; Martinez, A.; Mattei, M.G.; Kahn, A.; Raymondjean, M. Structure, sequence, and chromosomal location of the gene for USF2 transcription factors in mouse. Genomics 1995, 25, 36–43. [Google Scholar] [CrossRef]
- Henrion, A.A.; Vaulont, S.; Raymondjean, M.; Kahn, A. Mouse USF1 gene cloning: Comparative organization within the c-myc gene family. Mamm. Genome 1996, 7, 803–809. [Google Scholar] [CrossRef]
- Aperlo, C.; Boulukos, K.E.; Sage, J.; Cuzin, F.; Pognonec, P. Complete sequencing of the murine USF gene and comparison of its genomic organization to that of mFIP/USF2. Genomics 1996, 37, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Sirito, M.; Lin, Q.; Maity, T.; Sawadogo, M. Ubiquitous expression of the 43-and 44-kDa forms of transcription factor USF in mammalian cells. Nucleic Acids Res. 1994, 22, 427–433. [Google Scholar] [CrossRef]
- Sawadogo, M.; Dyke, M.W.V.; Gregor, P.D.; Roeder, R.G. Multiple forms of the human gene-specific transcription factor USF. I. Complete purification and identification of USF from HeLa cell nuclei. J. Biol. Chem. 1988, 263, 11985–11993. [Google Scholar]
- Yan, S.; Sloane, B.F. Isolation of a novel USF2 isoform: Repressor of cathepsin B expression. Gene 2004, 337, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Howcroft, T.K.; Murphy, C.; Weissman, J.D.; Huber, S.J.; Sawadogo, M.; Singer, D.S. Upstream stimulatory factor regulates major histocompatibility complex class I gene expression: The U2DeltaE4 splice variant abrogates E-box activity. Mol. Cell. Biol. 1999, 19, 4788–4797. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Ameur, A.; Kapranov, P.; Enroth, S.; Komorowski, J.; Gingeras, T.R.; Wadelius, C. Whole-genome maps of USF1 and USF2 binding and histone H3 acetylation reveal new aspects of promoter structure and candidate genes for common human disorders. Genome Res. 2008, 18, 380–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horbach, T.; Gotz, C.; Kietzmann, T.; Dimova, E.Y. Protein kinases as switches for the function of upstream stimulatory factors: Implications for tissue injury and cancer. Front. Pharm. 2015, 6, 3. [Google Scholar] [CrossRef]
- Dimova, E.Y.; Kietzmann, T. Cell type-dependent regulation of the hypoxia-responsive plasminogen activator inhibitor-1 gene by upstream stimulatory factor-2. J. Biol. Chem. 2006, 281, 2999–3005. [Google Scholar] [CrossRef]
- Samoylenko, A.; Dimova, E.Y.; Horbach, T.; Teplyuk, N.; Immenschuh, S.; Kietzmann, T. Opposite expression of the antioxidant heme oxygenase-1 in primary cells and tumor cells: Regulation by interaction of USF-2 and Fra-1. Antioxid. Redox Signal. 2008, 10, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Goueli, B.S.; Janknecht, R. Regulation of telomerase reverse transcriptase gene activity by upstream stimulatory factor. Oncogene 2003, 22, 8042–8047. [Google Scholar] [CrossRef] [Green Version]
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (hTERT) Gene. Genes (Basel) 2016, 7, 50. [Google Scholar] [CrossRef]
- Corre, S.; Galibert, M.D. Upstream stimulating factors: Highly versatile stress-responsive transcription factors. Pigment Cell Res. 2005, 18, 337–348. [Google Scholar] [CrossRef]
- Shimomura, K.; Kumar, V.; Koike, N.; Kim, T.; Chong, J.; Buhr, E.D.; Whiteley, A.R.; Low, S.S.; Omura, C.; Fenner, D.; et al. Usf1, a suppressor of the circadian Clock mutant, reveals the nature of the DNA-binding of the CLOCK:BMAL1 complex in mice. eLife 2013, 2013, e00426. [Google Scholar] [CrossRef]
- Vallet, V.S.; Henrion, A.A.; Bucchini, D.; Casado, M.; Raymondjean, M.; Kahn, A.; Vaulont, S. Glucose-dependent liver gene expression in upstream stimulatory factor 2−/− mice. J. Biol. Chem. 1997, 272, 21944–21949. [Google Scholar] [CrossRef]
- Chen, N.; Szentirmay, M.N.; Pawar, S.A.; Sirito, M.; Wang, J.; Wang, Z.; Zhai, Q.; Yang, H.X.; Peehl, D.M.; Ware, J.L.; et al. Tumor-suppression function of transcription factor USF2 in prostate carcinogenesis. Oncogene 2005, 25, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Ismail, P.M.; Lu, T.; Sawadogo, M. Loss of USF transcriptional activity in breast cancer cell lines. Oncogene 1999, 18, 5582–5591. [Google Scholar] [CrossRef] [Green Version]
- Szentirmay, M.N.; Yang, H.X.; Pawar, S.A.; Vinson, C.; Sawadogo, M. The IGF2 receptor is a USF2-specific target in nontumorigenic mammary epithelial cells but not in breast cancer cells. J. Biol. Chem. 2003, 278, 37231–37240. [Google Scholar] [CrossRef]
- Chang, J.T.C.; Yang, H.T.; Wang, T.C.V.; Cheng, A.J. Upstream stimulatory factor (USF) as a transcriptional suppressor of human telomerase reverse transcriptase (hTERT) in oral cancer cells. Mol. Carcinog. 2005, 44, 183–192. [Google Scholar] [CrossRef]
- Ocejo-Garcia, M.; Baokbah, T.A.; Ashurst, H.L.; Cowlishaw, D.; Soomro, I.; Coulson, J.M.; Woll, P.J. Roles for USF-2 in lung cancer proliferation and bronchial carcinogenesis. J. Pathol. 2005, 206, 151–159. [Google Scholar]
- Chen, B.; Chen, X.P.; Wu, M.S.; Cui, W.; Zhong, M. Expressions of heparanase and upstream stimulatory factor in hepatocellular carcinoma. Eur. J. Med. Res. 2014, 19, 45. [Google Scholar] [CrossRef]
- Tan, Y.; Chen, Y.; Du, M.; Peng, Z.; Xie, P. USF2 inhibits the transcriptional activity of Smurf1 and Smurf2 to promote breast cancer tumorigenesis. Cell Signal. 2019, 53, 49–58. [Google Scholar] [CrossRef]
- Sayasith, K.; Lussier, J.G.; Sirois, J. Role of upstream stimulatory factor phosphorylation in the regulation of the prostaglandin G/H synthase-2 promoter in granulosa cells. J. Biol. Chem. 2005, 280, 28885–28893. [Google Scholar] [CrossRef]
- Horbach, T.; Chi, T.F.; Gotz, C.; Sharma, S.; Juffer, A.H.; Dimova, E.Y.; Kietzmann, T. GSK3beta-dependent phosphorylation alters DNA binding, transactivity and half-life of the transcription factor USF2. PLoS ONE 2014, 9, e107914. [Google Scholar] [CrossRef]
- Lin, H.; Juang, J.L.; Wang, P.S. Involvement of Cdk5/p25 in digoxin-triggered prostate cancer cell apoptosis. J. Biol. Chem. 2004, 279, 29302–29307. [Google Scholar] [CrossRef]
- Strock, C.J.; Park, J.I.; Nakakura, E.K.; Bova, G.S.; Isaacs, J.T.; Ball, D.W.; Nelkin, B.D. Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Res. 2006, 66, 7509–7515. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.N.; Chen, M.C.; Chiang, M.C.; Lin, E.; Lee, Y.T.; Huang, P.H.; Lee, G.S.; Lin, H. Regulation of androgen receptor and prostate cancer growth by cyclin-dependent kinase 5. J. Biol. Chem. 2011, 286, 33141–33149. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, J.; Imanishi, S.Y.; Torvaldson, E.; Malinen, M.; Remes, M.; Orn, F.; Palvimo, J.J.; Eriksson, J.E. Cyclin-dependent kinase 5 acts as a critical determinant of AKT-dependent proliferation and regulates differential gene expression by the androgen receptor in prostate cancer cells. Mol. Biol. Cell 2015, 26, 1971–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.W.; Chang, J.G.; Yeh, K.T.; Chen, R.M.; Tsai, J.J.; Hu, R.M. Decreased expression of p39 is associated with a poor prognosis in human hepatocellular carcinoma. Med. Oncol. 2011, 28 (Suppl. 1), S239–S245. [Google Scholar] [CrossRef]
- Ehrlich, S.M.; Liebl, J.; Ardelt, M.A.; Lehr, T.; De Toni, E.N.; Mayr, D.; Brandl, L.; Kirchner, T.; Zahler, S.; Gerbes, A.L.; et al. Targeting cyclin dependent kinase 5 in hepatocellular carcinoma—A novel therapeutic approach. J. Hepatol. 2015, 63, 102–113. [Google Scholar] [CrossRef]
- Herzog, J.; Ehrlich, S.M.; Pfitzer, L.; Liebl, J.; Frohlich, T.; Arnold, G.J.; Mikulits, W.; Haider, C.; Vollmar, A.M.; Zahler, S. Cyclin-dependent kinase 5 stabilizes hypoxia-inducible factor-1alpha: A novel approach for inhibiting angiogenesis in hepatocellular carcinoma. Oncotarget 2016, 7, 27108–27121. [Google Scholar] [CrossRef]
- Zhang, R.; Lin, P.; Yang, H.; He, Y.; Dang, Y.W.; Feng, Z.B.; Chen, G. Clinical role and biological function of CDK5 in hepatocellular carcinoma: A study based on immunohistochemistry, RNA-seq and in vitro investigation. Oncotarget 2017, 8, 108333–108354. [Google Scholar] [CrossRef] [Green Version]
- Goodyear, S.; Sharma, M.C. Roscovitine regulates invasive breast cancer cell (MDA-MB231) proliferation and survival through cell cycle regulatory protein cdk5. Exp. Mol. Pathol. 2007, 82, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Li, L.; Zhang, J.; Lei, Y.; Wang, L.; Liu, D.X.; Feng, J.; Hou, P.; Yao, R.; Zhang, Y.; et al. CDK5 is essential for TGF-beta1-induced epithelial-mesenchymal transition and breast cancer progression. Sci. Rep. 2013, 3, 2932. [Google Scholar] [CrossRef] [PubMed]
- Chiker, S.; Pennaneach, V.; Loew, D.; Dingli, F.; Biard, D.; Cordelieres, F.P.; Gemble, S.; Vacher, S.; Bieche, I.; Hall, J.; et al. Cdk5 promotes DNA replication stress checkpoint activation through RPA-32 phosphorylation, and impacts on metastasis free survival in breast cancer patients. Cell Cycle 2015, 14, 3066–3078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozo, K.; Bibb, J.A. The Emerging Role of Cdk5 in Cancer. Trends Cancer 2016, 2, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Amanchy, R.; Periaswamy, B.; Mathivanan, S.; Reddy, R.; Tattikota, S.G.; Pandey, A. A curated compendium of phosphorylation motifs. Nat. Biotechnol. 2007, 25, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Gnad, F.; Ren, S.; Cox, J.; Olsen, J.V.; Macek, B.; Oroshi, M.; Mann, M. PHOSIDA (phosphorylation site database): Management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 2007, 8, R250. [Google Scholar] [CrossRef]
- Löffler, I.; Grün, M.; Böhmer, F.D.; Rubio, I. Role of cAMP in the promotion of colorectal cancer cell growth by Prostaglandin E2. BMC Cancer 2008, 8, 380. [Google Scholar] [CrossRef]
- Porter, S.E.; Dwyer-Nield, L.D.; Malkinson, A.M. Regulation of lung epithelial cell morphology by cAMP-dependent protein kinase type I isozyme. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L1282–L1289. [Google Scholar] [CrossRef]
- Landa, I.; Ruiz-Llorente, S.; Montero-Conde, C.; Inglada-Perez, L.; Schiavi, F.; Leskela, S.; Pita, G.; Milne, R.; Maravall, J.; Ramos, I.; et al. The variant rs1867277 in FOXE1 gene confers thyroid cancer susceptibility through the recruitment of USF1/USF2 transcription factors. PLoS Genet. 2009, 5, e1000637. [Google Scholar] [CrossRef] [PubMed]
- Groenen, P.; Garcia, E.; Debeer, P.; Devriendt, K.; Fryns, J.P.; Ven, W.J.M. Structure, sequence, and chromosome 19 localization of human USF2 and its rearrangement in a patient with multicystic renal dysplasia. Genomics 1996, 38, 141–148. [Google Scholar] [CrossRef]
- Luo, X.; Sawadogo, M. Functional domains of the transcription factor USF2: Atypical nuclear localization signals and context-dependent transcriptional activation domains. Mol. Cell. Biol. 1996, 16, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Qyang, Y.; Luo, X.; Lu, T.; Ismail, P.M.; Krylov, D.; Vinson, C.; Sawadogo, M. Cell-type-dependent activity of the ubiquitous transcription factor USF in cellular proliferation and transcriptional activation. Mol. Cell. Biol. 1999, 19, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Beaudette, K.N.; Lew, J.; Wang, J.H. Substrate specificity characterization of a cdc2-like protein kinase purified from bovine brain. J. Biol. Chem. 1993, 268, 20825–20830. [Google Scholar] [PubMed]
- Shetty, K.T.; Link, W.T.; Pant, H.C. cdc2-like kinase from rat spinal cord specifically phosphorylates KSPXK motifs in neurofilament proteins: Isolation and characterization. Proc. Natl. Acad. Sci. USA 1993, 90, 6844–6848. [Google Scholar] [CrossRef] [PubMed]
- Songyang, Z.; Lu, K.P.; Kwon, Y.T.; Tsai, L.H.; Filhol, O.; Cochet, C.; Brickey, D.A.; Soderling, T.R.; Bartleson, C.; Graves, D.J.; et al. A structural basis for substrate specificities of protein Ser/Thr kinases: Primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol. Cell. Biol. 1996, 16, 6486–6493. [Google Scholar] [CrossRef]
- Kamei, H.; Saito, T.; Ozawa, M.; Fujita, Y.; Asada, A.; Bibb, J.A.; Saido, T.C.; Sorimachi, H.; Hisanaga, S. Suppression of calpain-dependent cleavage of the CDK5 activator p35 to p25 by site-specific phosphorylation. J. Biol. Chem. 2007, 282, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, H.S.; Lee, S.J.; Kim, K.T. Stabilization and activation of p53 induced by Cdk5 contributes to neuronal cell death. J. Cell Sci. 2007, 120, 2259–2271. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Zhou, J.; Zhang, J.; Wu, S.; Yang, X.; Zhao, X.; Li, H.; Luo, M.; Yu, Q.; Lin, G.; et al. Cyclin-dependent kinase 5 decreases in gastric cancer and its nuclear accumulation suppresses gastric tumorigenesis. Clin. Cancer Res. 2015, 21, 1419–1428. [Google Scholar] [CrossRef]
- Xu, S.; Li, X.; Gong, Z.; Wang, W.; Li, Y.; Nair, B.C.; Piao, H.; Yang, K.; Wu, G.; Chen, J. Proteomic analysis of the human cyclin-dependent kinase family reveals a novel CDK5 complex involved in cell growth and migration. Mol. Cell. Proteom. 2014, 13, 2986–3000. [Google Scholar] [CrossRef]
- Van den Heuvel, S.; Harlow, E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science 1993, 262, 2050–2054. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chi, T.F.; Horbach, T.; Götz, C.; Kietzmann, T.; Dimova, E.Y. Cyclin-Dependent Kinase 5 (CDK5)-Mediated Phosphorylation of Upstream Stimulatory Factor 2 (USF2) Contributes to Carcinogenesis. Cancers 2019, 11, 523. https://doi.org/10.3390/cancers11040523
Chi TF, Horbach T, Götz C, Kietzmann T, Dimova EY. Cyclin-Dependent Kinase 5 (CDK5)-Mediated Phosphorylation of Upstream Stimulatory Factor 2 (USF2) Contributes to Carcinogenesis. Cancers. 2019; 11(4):523. https://doi.org/10.3390/cancers11040523
Chicago/Turabian StyleChi, Tabughang Franklin, Tina Horbach, Claudia Götz, Thomas Kietzmann, and Elitsa Y. Dimova. 2019. "Cyclin-Dependent Kinase 5 (CDK5)-Mediated Phosphorylation of Upstream Stimulatory Factor 2 (USF2) Contributes to Carcinogenesis" Cancers 11, no. 4: 523. https://doi.org/10.3390/cancers11040523
APA StyleChi, T. F., Horbach, T., Götz, C., Kietzmann, T., & Dimova, E. Y. (2019). Cyclin-Dependent Kinase 5 (CDK5)-Mediated Phosphorylation of Upstream Stimulatory Factor 2 (USF2) Contributes to Carcinogenesis. Cancers, 11(4), 523. https://doi.org/10.3390/cancers11040523