Transient Receptor Potential Mucolipin-1 Channels in Glioblastoma: Role in Patient’s Survival




 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. TRPML-1 Expression in T98 and U251 GBM Cell Lines
2.2. Subcellular Expression of TRPML-1 in Glioblastoma Cell Lines
2.3. The Specific TRPML-1 Agonist, MK6-83, Triggers Intracellular Ca2+ Rise and Inhibits the Viability in T98 and U251 Cells
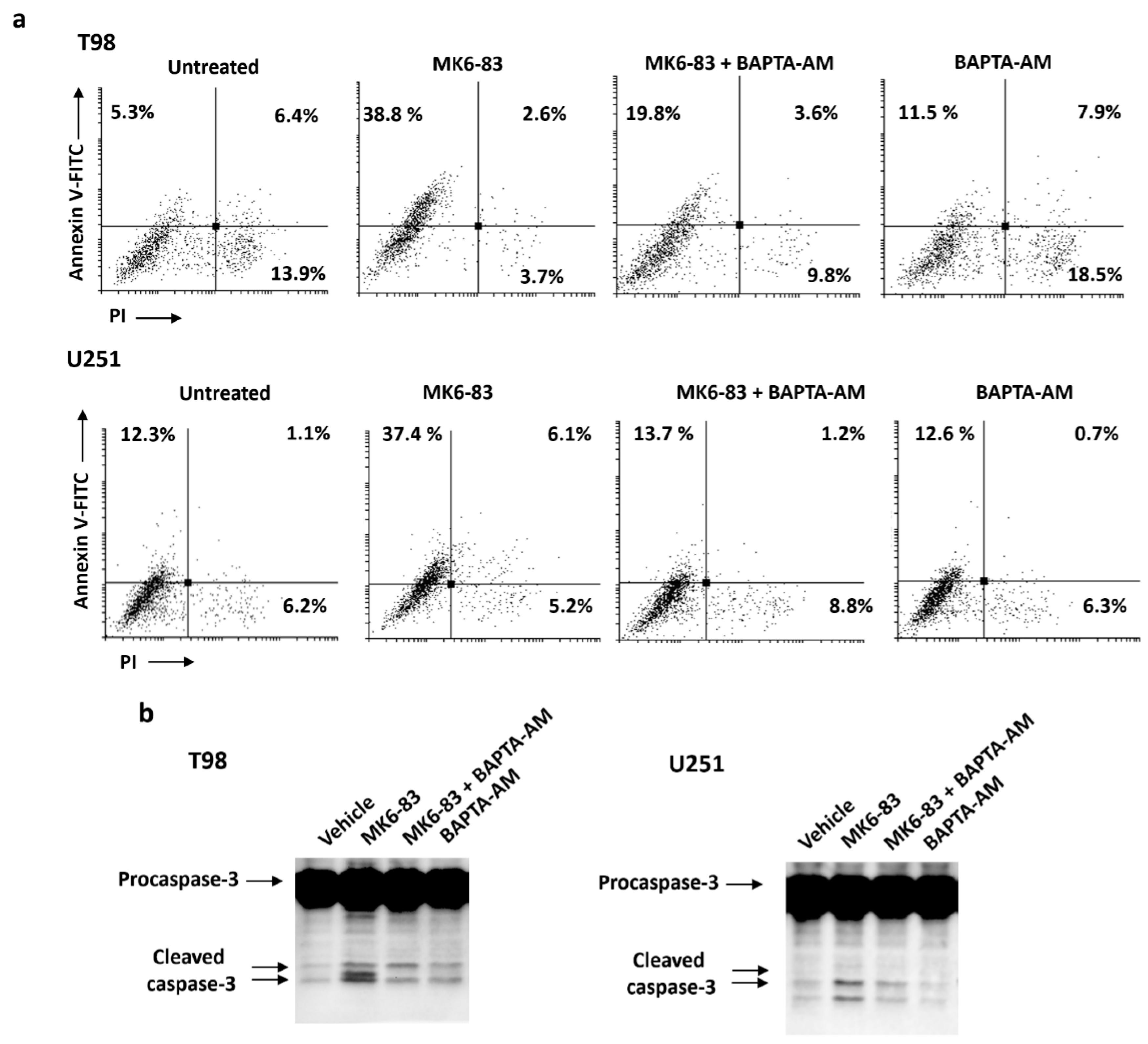
2.4. TRPML-1 Activation Triggers Caspase-Dependent Apoptosis in T98 and U251 Cells
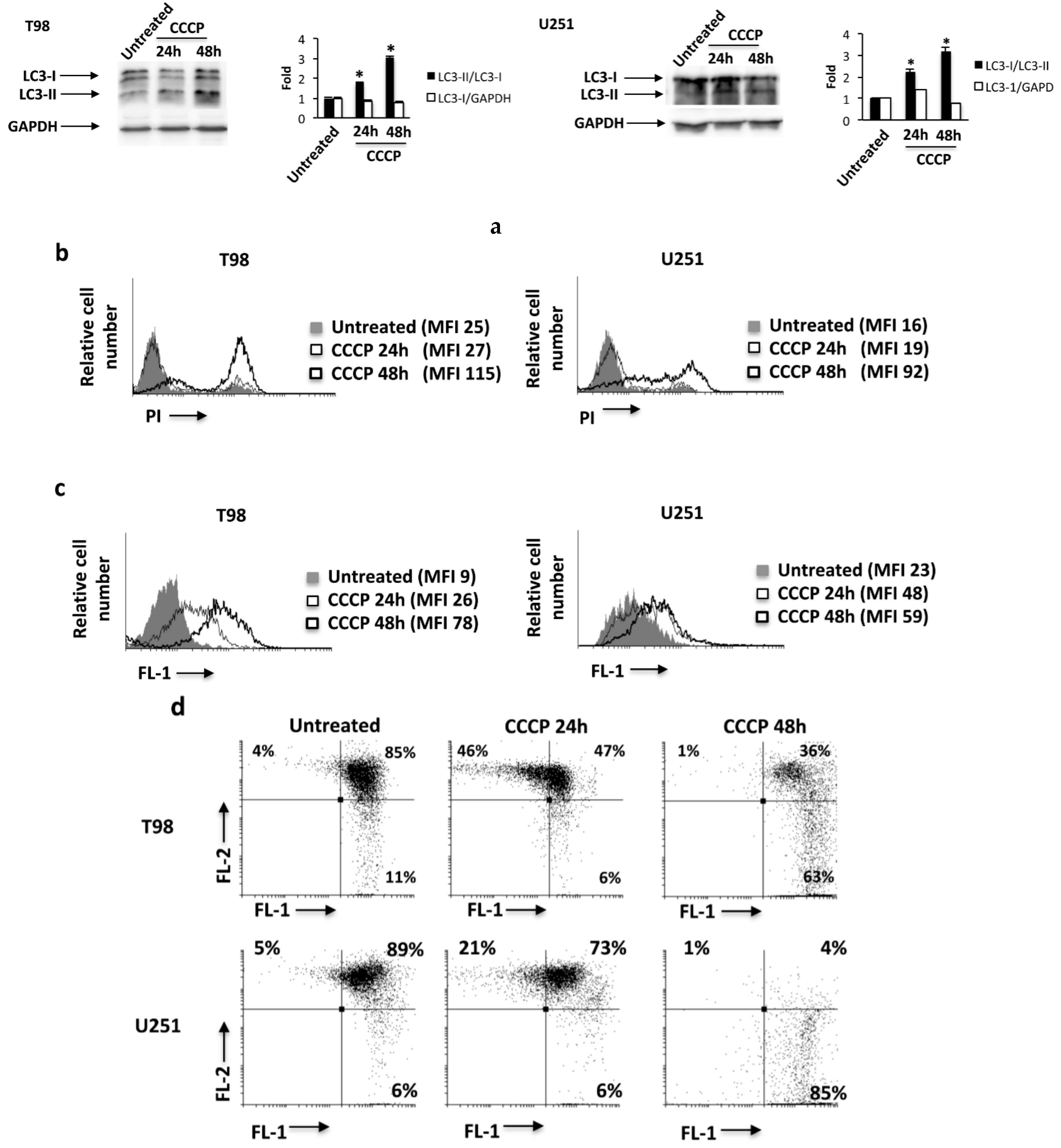
2.5. The ROS Inducer, Carbonyl Cyanide m-Chlorophenylhydrazone (CCCP), Triggers TRPML-1-Dependent Autophagic Cell Death in GBM Cell Lines
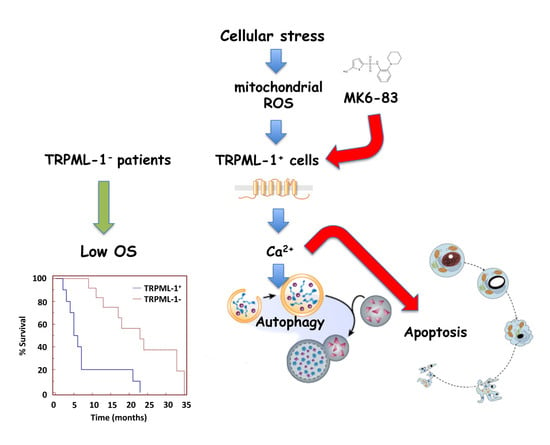
2.6. TRPML-1 as Prognostic Factor in GBM Patients
3. Discussion
4. Materials and Methods
4.1. Cells and Tissues
4.2. Chemical and Reagents
4.3. Western Blot Analysis
4.4. Protein-DNA Binding Assay
4.5. TRPML-1 Transfection Models
4.6. MTT Assay
4.7. Calcium Mobilization Assay
4.8. Cell Cycle Analysis
4.9. Mitochondrial Transmembrane Potential (ΔΨm)
4.10. ROS Production
4.11. Cell Death Analysis
4.12. Gene Expression Analysis
4.13. Immunohistochemistry
4.14. Confocal Laser Scanning Microscopy Analysis
4.15. Immunofluorescence and FACS Analysis
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef]
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 215–225. [Google Scholar] [CrossRef]
- Santoni, G.; Morelli, M.B.; Amantini, C.; Santoni, M.; Nabissi, M. New insight on the role of Transient Receptor Potential (TRP) channels in driven gliomagenesis pathways. In GLIOMA Exploring Its Biology and Practical Relevance; Ghosh, A., Ed.; Intech: Rijeka, Croatia, 2011; pp. 163–188. [Google Scholar]
- Liberati, S.; Morelli, M.B.; Nabissi, M.; Santoni, M.; Santoni, G. Oncogenic and anti-oncogenic effects of transient receptor potential channels. Curr. Top. Med. Chem. 2013, 13, 344–366. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Tomassoni, D.; Rossi, F.; Cardinali, C.; Santoni, M.; Arcella, A.; Oliva, M.A.; Santoni, A.; et al. Overexpression of transient receptor potential mucolipin-2 ion channels in gliomas: Role in tumor growth and progression. Oncotarget 2016, 7, 43654–43668. [Google Scholar] [CrossRef]
- Acierno, J.S., Jr.; Kennedy, J.C.; Falardeau, J.L.; Leyne, M.; Bromley, M.C.; Colman, M.W.; Sun, M.; Bove, C.; Ashworth, L.K.; Chadwick, L.H.; et al. A physical and transcript map of the MCOLN1 gene region on human chromosome 19p13.3-p13.2. Genomics 2001, 73, 203–210. [Google Scholar] [CrossRef]
- Bargal, R.; Avidan, N.; Ben-Asher, E.; Olender, Z.; Zeigler, M.; Frumkin, A.; Raas-Rothschild, A.; Glusman, G.; Lancet, D.; Bach, G. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 2000, 26, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Goldin, E.; Stahl, S.; Falardeau, J.L.; Kennedy, J.C.; Acierno, J.S., Jr.; Bove, C.; Kaneski, C.R.; Nagle, J.; Bromley, M.C.; et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 2000, 9, 2471–2478. [Google Scholar] [CrossRef] [Green Version]
- Slaugenhaupt, S.A. The molecular basis of mucolipidosis type IV. Curr. Mol. Med. 2002, 2, 445–450. [Google Scholar] [CrossRef]
- Chen, Q.; She, J.; Zeng, W.; Guo, J.; Xu, H.; Bai, X.C.; Jiang, Y. Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature 2017, 550, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Pryor, P.R.; Reimann, F.; Gribble, F.M.; Luzio, J.P. Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic 2006, 7, 1388–1398. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Hofmann, T.; Montell, C. Lysosomal localization of TRPML3 dependes on TRPML2 and the mucolipidosis-associated protein TRPML1. J. Biol. Chem. 2006, 281, 17517–17527. [Google Scholar] [CrossRef]
- Li, M.; Zhang, W.K.; Benvin, N.M.; Zhou, X.; Su, D.; Li, H.; Wang, S.; Michailidis, I.E.; Tong, L.; Li, X.; et al. Structural basis of dual Ca2+/pH regulation of the endolysosomal TRPML1 channel. Nat. Struct. Mol. Biol. 2017, 24, 205–213. [Google Scholar] [CrossRef]
- Venugopal, B.; Mesires, N.T.; Kennedy, J.C.; Curcio-Morelli, C.; Laplante, J.M.; Dice, J.F.; Slaugenhaupt, S.A. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J. Cell. Physiol. 2009, 219, 344–353. [Google Scholar] [CrossRef]
- Vergarajauregui, S.; Martina, J.A.; Puertollano, R. Identification of the penta-EF-hand protein ALG-2 as a Ca2+-dependent interactor of mucolipin-1. J. Biol. Chem. 2009, 284, 36357–36366. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, F.; Pang, Y.; Zhao, E.; Zhu, S.; Chen, F.; Cui, H. ALG2 regulates glioma cell proliferation, migration and tumorigenicity. Biochem. Biophys. Res. Commun. 2017, 486, 300–306. [Google Scholar] [CrossRef]
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef]
- Chen, C.C.; Keller, M.; Hess, M.; Schiffmann, R.; Urban, N.; Wolfgardt, A.; Schaefer, M.; Bracher, F.; Biel, M.; Wahl-Schott, C.; et al. A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat. Commun. 2014, 5, 4681. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef]
- Zhang, X.; Li, X.; Xu, H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc. Natl. Acad. Sci. USA 2012, 109, 11384–11389. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Delling, M.; Li, L.; Dong, X.; Clapham, D.E. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc. Natl. Acad. Sci. USA 2007, 104, 18321–18326. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.Z.; Zou, Y.; Sun, X.; Dong, G.; Cao, Q.; Pandey, A.; Rainey, J.K.; Zhu, X.; Dong, X.P. Inhibition of Transient Receptor Potential Channel Mucolipin-1 (TRPML1) by Lysosomal Adenosine Involved in Severe Combined Immunodeficiency Diseases. J. Biol. Chem. 2017, 292, 3445–3455. [Google Scholar] [CrossRef]
- Onyenwoke, R.U.; Sexton, J.Z.; Yan, F.; Díaz, M.C.; Forsberg, L.J.; Major, M.B.; Brenman, J.E. The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem. J. 2015, 470, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, S.; Wu, S.; Zhang, J.; Nie, Z.; Zeng, S. A novel role of transient receptor potential mucolipin1 (TRPML1) in protecting against imidazole-induced cytotoxicity. Biochem. Cell Biol. 2014, 92, 279–286. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [Green Version]
- Curcio-Morelli, C.; Zhang, P.; Venugopal, B.; Charles, F.A.; Browning, M.F.; Cantiello, H.F.; Slaugenhaupt, S.A. Functional multimerization of mucolipin channel proteins. J. Cell. Physiol. 2010, 222, 328–335. [Google Scholar] [CrossRef]
- Grimm, C.; Hassan, S.; Wahl-Schott, C.; Biel, M. Role of TRPML and two pore channels in endolysosomal cation homeostasis. J. Pharmacol. Exp. Ther. 2012, 342, 236–244. [Google Scholar] [CrossRef]
- Zeevi, D.A.; Lev, S.; Frumkin, A.; Minke, B.; Bach, G. Heteromultimeric TRPML channel assemblies play a crucial role in the regulation of cell viability models and starvation-induced autophagy. J. Cell Sci. 2010, 123, 3112–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, S.; Jha, A.; Li, Q.; Soyombo, A.A.; Dickinson, G.D.; Churamani, D.; Brailoiu, E.; Patel, S.; Muallem, S. Transient receptor potential mucolipin 1 (TRPML1) and two-pore channels are functionally independent organellar ion channels. J. Biol. Chem. 2011, 286, 22934–22942. [Google Scholar] [CrossRef]
- Grimm, C.; Jörs, S.; Saldanha, S.A.; Obukhov, A.G.; Pan, B.; Oshima, K.; Cuajungco, M.P.; Chase, P.; Hodder, P.; Heller, S. Small molecule activators of TRPML3. Chem. Biol. 2010, 17, 135–148. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, L.; Xu, H. Lysosome calcium in ROS regulation of autophagy. Autophagy 2016, 12, 1954–1955. [Google Scholar] [CrossRef] [Green Version]
- Grimm, C.; Bartel, K.; Vollmar, A.M.; Biel, M. Endolysosomal cation channels and cancer-A link with great potential. Pharmaceuticals 2018, 11, 4. [Google Scholar] [CrossRef]
- Zeng, X.; Sikka, S.C.; Huang, L.; Sun, C.; Xu, C.; Jia, D.; Abdel-Mageed, A.B.; Pottle, J.E.; Taylor, J.T.; Li, M. Novel role for the transient receptor potential channel TRPM2 in prostate cancer cell proliferation. Prostate Cancer Prostatic Dis. 2010, 13, 195–201. [Google Scholar] [CrossRef]
- Hopkins, M.M.; Feng, X.; Liu, M.; Parker, L.P.; Koh, D.W. Inhibition of the transient receptor potential melastatin-2 channel causes increased DNA damage and decreased proliferation in breast adenocarcinoma cells. Int. J. Oncol. 2015, 46, 2267–2276. [Google Scholar] [CrossRef] [Green Version]
- McLane, L.M.; Corbett, A.H. Nuclear localization signals and human disease. IUBMB Life 2009, 61, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Ezak, M.J.; Ferkey, D.M. A functional nuclear localization sequence in the C. elegans TRPV channel OCR-2. PLoS ONE 2011, 6, e25047. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Medina, D.L.; Ballabio, A. Lysosomal calcium regulates autophagy. Autophagy 2015, 11, 970–971. [Google Scholar] [CrossRef] [Green Version]
- East, D.A.; Campanella, M. Ca2+ in quality control: An unresolved riddle critical to autophagy and mitophagy. Autophagy 2013, 9, 1710–1719. [Google Scholar] [CrossRef]
- De Maria, R.; Palmieri, G.; Santoni, A. Induction of Ca2+ flux by adhesion molecules in lymphocytes. Methods Mol. Biol. 1999, 96, 199–204. [Google Scholar]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morelli, M.B.; Amantini, C.; Tomassoni, D.; Nabissi, M.; Arcella, A.; Santoni, G. Transient Receptor Potential Mucolipin-1 Channels in Glioblastoma: Role in Patient’s Survival. Cancers 2019, 11, 525. https://doi.org/10.3390/cancers11040525
Morelli MB, Amantini C, Tomassoni D, Nabissi M, Arcella A, Santoni G. Transient Receptor Potential Mucolipin-1 Channels in Glioblastoma: Role in Patient’s Survival. Cancers. 2019; 11(4):525. https://doi.org/10.3390/cancers11040525
Chicago/Turabian StyleMorelli, Maria Beatrice, Consuelo Amantini, Daniele Tomassoni, Massimo Nabissi, Antonella Arcella, and Giorgio Santoni. 2019. "Transient Receptor Potential Mucolipin-1 Channels in Glioblastoma: Role in Patient’s Survival" Cancers 11, no. 4: 525. https://doi.org/10.3390/cancers11040525
APA StyleMorelli, M. B., Amantini, C., Tomassoni, D., Nabissi, M., Arcella, A., & Santoni, G. (2019). Transient Receptor Potential Mucolipin-1 Channels in Glioblastoma: Role in Patient’s Survival. Cancers, 11(4), 525. https://doi.org/10.3390/cancers11040525