Fibroblasts as Modulators of Local and Systemic Cancer Metabolism

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Wound-Healing Mediators as Metabolic Regulators
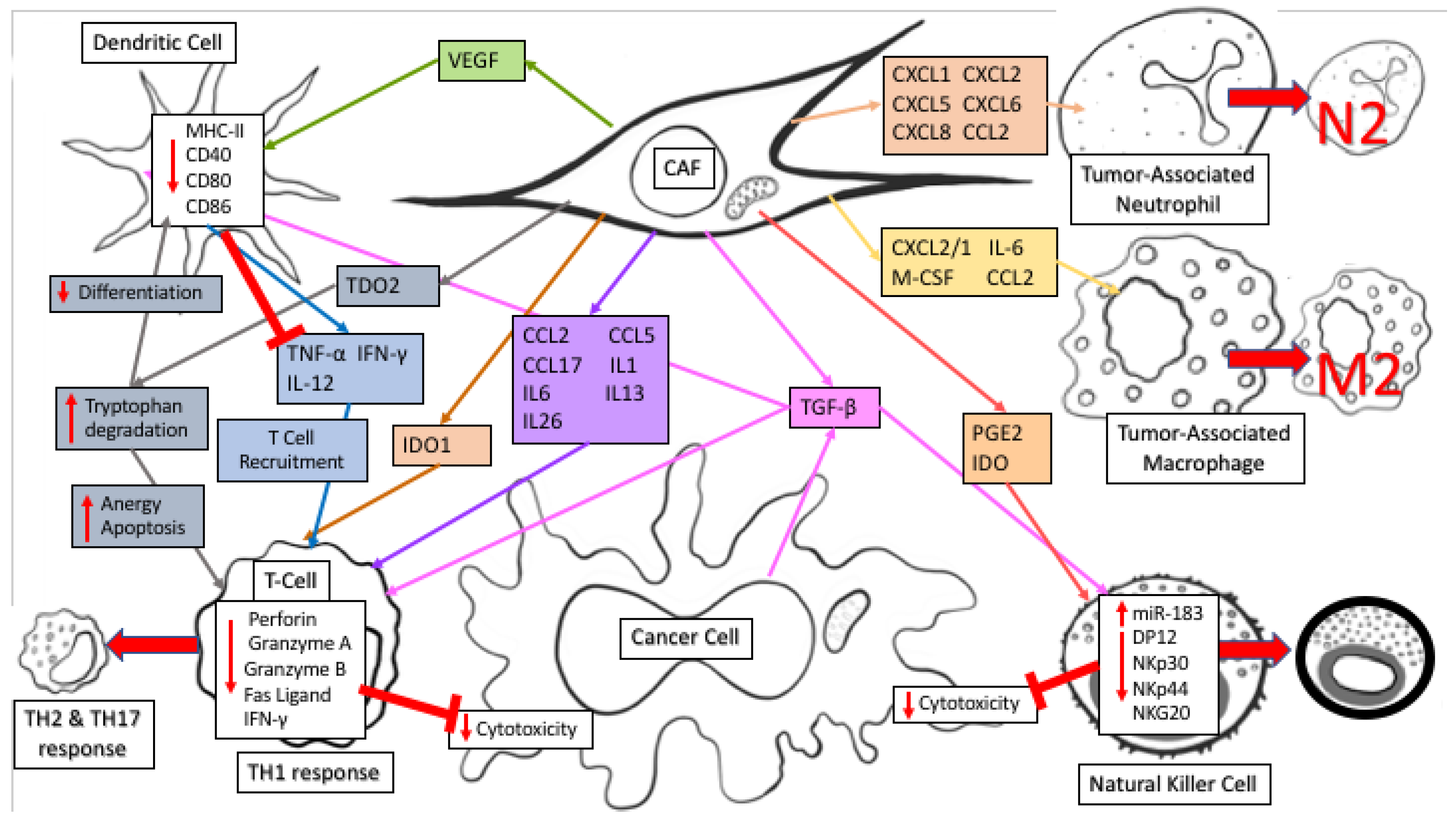
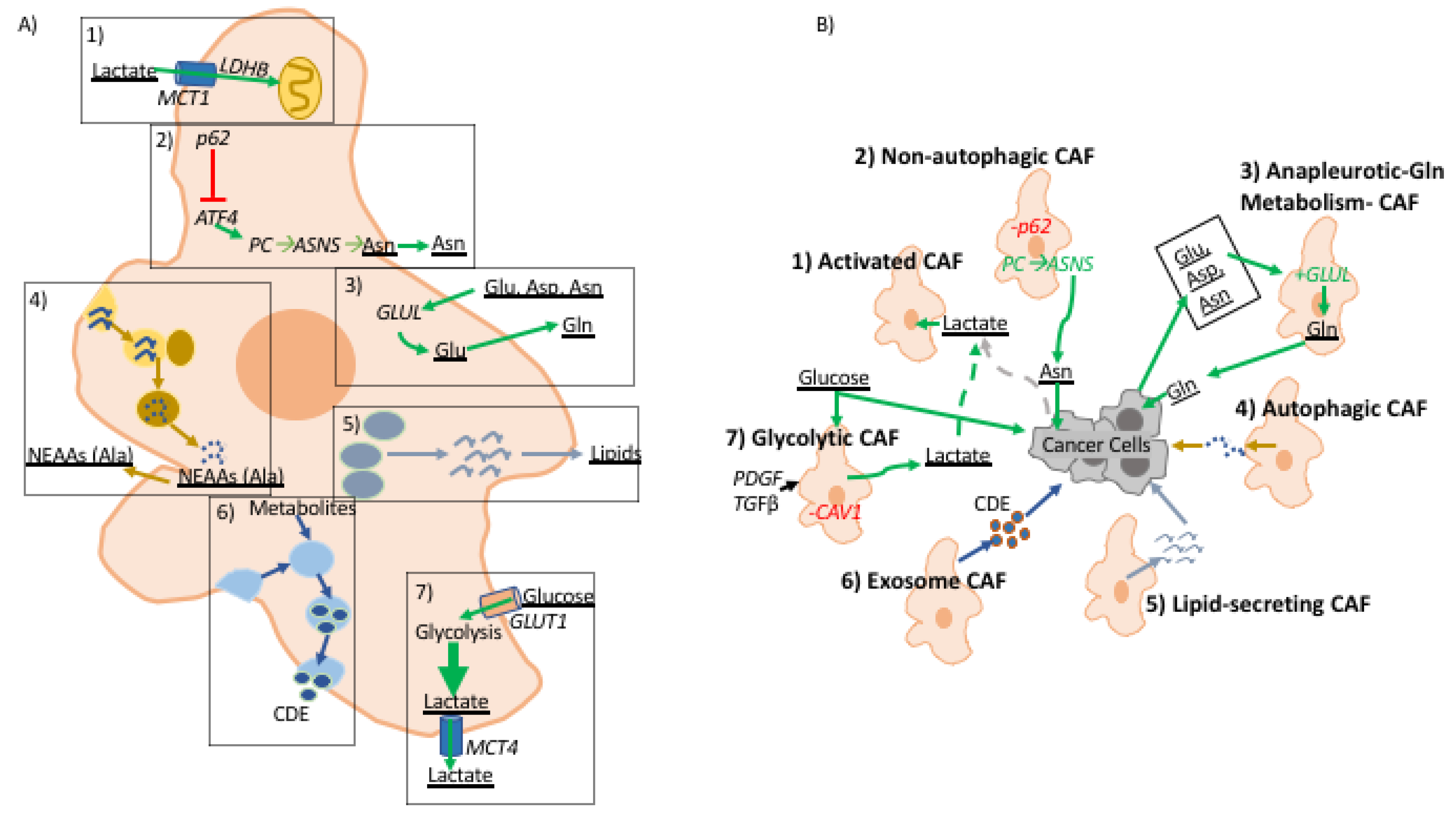
3. A Role for Fibroblast-Derived Metabolites in Tumor-Stroma Interaction
4. Fibroblasts as Determinants of Systemic Metabolism in Cancer
5. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Paunescu, V.; Bojin, F.M.; Tatu, C.A.; Gavriliuc, O.I.; Rosca, A.; Gruia, A.T.; Tanasie, G.; Bunu, C.; Crisnic, D.; Gherghiceanu, M.; et al. Tumour-associated fibroblasts and mesenchymal stem cells: More similarities than differences. J. Cell. Mol. Med. 2011, 15, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL-1-induced JAK/STAT signaling is antagonized by TGF-beta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2018. [Google Scholar] [CrossRef]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Celis, J.E.; Moreira, J.M.; Gromova, I.; Cabezon, T.; Ralfkiaer, U.; Guldberg, P.; Straten, P.T.; Mouridsen, H.; Friis, E.; Holm, D.; et al. Towards discovery-driven translational research in breast cancer. FEBS J. 2005, 272, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Chiarugi, P.; Cirri, P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016, 380, 272–280. [Google Scholar] [CrossRef]
- Miller, A.; Nagy, C.; Knapp, B.; Laengle, J.; Ponweiser, E.; Groeger, M.; Starkl, P.; Bergmann, M.; Wagner, O.; Haschemi, A. Exploring Metabolic Configurations of Single Cells within Complex Tissue Microenvironments. Cell Metab. 2017, 26, 788–800.e6. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Algul, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.N.; Berthezene, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.; Helou, Y.; Batlle, R.; Liu, X.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019, 29, 141–155.e9. [Google Scholar] [CrossRef]
- Sullivan, W.J.; Mullen, P.J.; Schmid, E.W.; Flores, A.; Momcilovic, M.; Sharpley, M.S.; Jelinek, D.; Whiteley, A.E.; Maxwell, M.B.; Wilde, B.R.; et al. Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell 2018, 175, 117–132.e121. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140.e10. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kryczek, I.; Dostal, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Hung, J.Y.; Chiang, S.Y.; Jian, S.F.; Wu, C.Y.; Lin, Y.S.; Tsai, Y.M.; Chou, S.H.; Tsai, M.J.; Kuo, P.L. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget 2016, 7, 27584–27598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H., 3rd; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 2016, 532, 250–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.N.; Vidal, N.; Berthezene, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Sotgia, F.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Lisanti, M.P. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011, 13, 213. [Google Scholar] [CrossRef]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of cancer-associated fibroblasts by IDH3alpha downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Fuyuhiro, Y.; Yashiro, M.; Noda, S.; Kashiwagi, S.; Matsuoka, J.; Doi, Y.; Kato, Y.; Hasegawa, T.; Sawada, T.; Hirakawa, K. Upregulation of cancer-associated myofibroblasts by TGF-beta from scirrhous gastric carcinoma cells. Br. J. Cancer 2011, 105, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Ando, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-beta drives tumor growth: Connecting TGF-beta signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Rattigan, Y.I.; Patel, B.B.; Ackerstaff, E.; Sukenick, G.; Koutcher, J.A.; Glod, J.W.; Banerjee, D. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp. Cell Res. 2012, 318, 326–335. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzola, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Valencia, T.; Kim, J.Y.; Abu-Baker, S.; Moscat-Pardos, J.; Ahn, C.S.; Reina-Campos, M.; Duran, A.; Castilla, E.A.; Metallo, C.M.; Diaz-Meco, M.T.; et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef]
- Linares, J.F.; Cordes, T.; Duran, A.; Reina-Campos, M.; Valencia, T.; Ahn, C.S.; Castilla, E.A.; Moscat, J.; Metallo, C.M.; Diaz-Meco, M.T. ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab. 2017, 26, 817–829.e6. [Google Scholar] [CrossRef]
- Sherman, M.H. Stellate Cells in Tissue Repair, Inflammation, and Cancer. Annu. Rev. Cell Dev. Biol. 2018, 34, 333–355. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nardi, F.; Fitchev, P.; Franco, O.E.; Ivanisevic, J.; Scheibler, A.; Hayward, S.W.; Brendler, C.B.; Welte, M.A.; Crawford, S.E. PEDF regulates plasticity of a novel lipid-MTOC axis in prostate cancer-associated fibroblasts. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Guillaumond, F. LDL Receptor: An open route to feed pancreatic tumor cells. Mol. Cell Oncol 2016, 3, e1033586. [Google Scholar] [CrossRef]
- Garcia-Bermudez, J.; Baudrier, L.; Bayraktar, E.C.; Shen, Y.; La, K.; Guarecuco, R.; Yucel, B.; Fiore, D.; Tavora, B.; Freinkman, E.; et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 2019, 567, 118–122. [Google Scholar] [CrossRef]
- Gangoda, L.; Boukouris, S.; Liem, M.; Kalra, H.; Mathivanan, S. Extracellular vesicles including exosomes are mediators of signal transduction: Are they protective or pathogenic? Proteomics 2015, 15, 260–271. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Wagner, E.F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 2016, 30, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Mayers, J.R.; Wu, C.; Clish, C.B.; Kraft, P.; Torrence, M.E.; Fiske, B.P.; Yuan, C.; Bao, Y.; Townsend, M.K.; Tworoger, S.S.; et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014, 20, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, M.; Haji, S.; Amagai, T. High Serum Essential Amino Acids as a Predictor of Skeletal Muscle Depletion in Patients With Cachexia and Advanced Gastrointestinal Cancers. Nutr. Clin. Pract. 2017, 32, 645–651. [Google Scholar] [CrossRef]
- Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. USA 1990, 87, 7235–7239. [Google Scholar] [CrossRef]
- Brokopp, C.E.; Schoenauer, R.; Richards, P.; Bauer, S.; Lohmann, C.; Emmert, M.Y.; Weber, B.; Winnik, S.; Aikawa, E.; Graves, K.; et al. Fibroblast activation protein is induced by inflammation and degrades type I collagen in thin-cap fibroatheromata. Eur. Heart J. 2011, 32, 2713–2722. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.; et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef] [Green Version]
- Argiles, J.M.; Busquets, S.; Lopez-Soriano, F.J. Cytokines in the pathogenesis of cancer cachexia. Curr Opin Clin. Nutr. Metab. Care 2003, 6, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Ohe, Y.; Podack, E.R.; Olsen, K.J.; Miyahara, Y.; Miura, K.; Saito, H.; Koishihara, Y.; Ohsugi, Y.; Ohira, T.; Nishio, K.; et al. Interleukin-6 cDNA transfected Lewis lung carcinoma cells show unaltered net tumour growth rate but cause weight loss and shortened survival in syngeneic mice. Br. J. Cancer 1993, 67, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujinaka, T.; Ebisui, C.; Fujita, J.; Kishibuchi, M.; Morimoto, T.; Ogawa, A.; Katsume, A.; Ohsugi, Y.; Kominami, E.; Monden, M. Muscle undergoes atrophy in association with increase of lysosomal cathepsin activity in interleukin-6 transgenic mouse. Biochem. Biophys Res. Commun. 1995, 207, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Black, K.; Garrett, I.R.; Mundy, G.R. Chinese hamster ovarian cells transfected with the murine interleukin-6 gene cause hypercalcemia as well as cachexia, leukocytosis and thrombocytosis in tumor-bearing nude mice. Endocrinology 1991, 128, 2657–2659. [Google Scholar] [CrossRef]
- Strassmann, G.; Fong, M.; Kenney, J.S.; Jacob, C.O. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J. Clin. Invest. 1992, 89, 1681–1684. [Google Scholar] [CrossRef]
- Strassmann, G.; Fong, M.; Freter, C.E.; Windsor, S.; D’Alessandro, F.; Nordan, R.P. Suramin interferes with interleukin-6 receptor binding in vitro and inhibits colon-26-mediated experimental cancer cachexia in vivo. J. Clin. Invest. 1993, 92, 2152–2159. [Google Scholar] [CrossRef]
- Tamura, S.; Ouchi, K.F.; Mori, K.; Endo, M.; Matsumoto, T.; Eda, H.; Tanaka, Y.; Ishitsuka, H.; Tokita, H.; Yamaguchi, K. Involvement of human interleukin 6 in experimental cachexia induced by a human uterine cervical carcinoma xenograft. Clin. Cancer Res. 1995, 1, 1353–1358. [Google Scholar]
- Staal-van den Brekel, A.J.; Dentener, M.A.; Schols, A.M.; Buurman, W.A.; Wouters, E.F. Increased resting energy expenditure and weight loss are related to a systemic inflammatory response in lung cancer patients. J. Clin. Oncol. 1995, 13, 2600–2605. [Google Scholar] [CrossRef]
- Fearon, K.C.; McMillan, D.C.; Preston, T.; Winstanley, F.P.; Cruickshank, A.M.; Shenkin, A. Elevated circulating interleukin-6 is associated with an acute-phase response but reduced fixed hepatic protein synthesis in patients with cancer. Ann. Surg. 1991, 213, 26–31. [Google Scholar] [CrossRef]
- Okada, S.; Okusaka, T.; Ishii, H.; Kyogoku, A.; Yoshimori, M.; Kajimura, N.; Yamaguchi, K.; Kakizoe, T. Elevated serum interleukin-6 levels in patients with pancreatic cancer. Jpn. J. Clin. Oncol. 1998, 28, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Aydogdu, T.; Kunzevitzky, N.; Guttridge, D.C.; Khuri, S.; Koniaris, L.G.; Zimmers, T.A. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One 2011, 6, e22538. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 2018, 37, 873–883. [Google Scholar] [CrossRef]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef] [Green Version]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, W.; Collins, M.A.; Bednar, F.; Rakshit, S.; Zetter, B.R.; Stanger, B.Z.; Chung, I.; Rhim, A.D.; di Magliano, M.P. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013, 73, 6359–6374. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dunn, A.J. Mouse interleukin-6 stimulates the HPA axis and increases brain tryptophan and serotonin metabolism. Neurochem. Int. 1998, 33, 143–154. [Google Scholar] [CrossRef]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Murata, T.; Mizushima, H.; Chinen, I.; Moribe, H.; Yagi, S.; Hoffman, R.M.; Kimura, T.; Yoshino, K.; Ueda, Y.; Enomoto, T.; et al. HB-EGF and PDGF mediate reciprocal interactions of carcinoma cells with cancer-associated fibroblasts to support progression of uterine cervical cancers. Cancer Res. 2011, 71, 6633–6642. [Google Scholar] [CrossRef]
- Neufert, C.; Becker, C.; Tureci, O.; Waldner, M.J.; Backert, I.; Floh, K.; Atreya, I.; Leppkes, M.; Jefremow, A.; Vieth, M.; et al. Tumor fibroblast-derived epiregulin promotes growth of colitis-associated neoplasms through ERK. J. Clin. Invest. 2013, 123, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Cros, M.; Cataisson, C.; Cho, Y.M.; Berthois, Y.; Bernard-Poenaru, O.; Denne, M.; Graulet, A.M.; De Vernejoul, M.C.; Foley, J.; Bouizar, Z. Constitutive production of parathyroid hormone-related protein (PTHrP) by fibroblasts derived from normal and pathological human breast tissue. Oncol. Res. 2002, 13, 137–146. [Google Scholar]
- Blomme, E.A.; Sugimoto, Y.; Lin, Y.C.; Capen, C.C.; Rosol, T.J. Parathyroid hormone-related protein is a positive regulator of keratinocyte growth factor expression by normal dermal fibroblasts. Mol. Cell Endocrinol. 1999, 152, 189–197. [Google Scholar] [CrossRef]
- Foley, J.; Nickerson, N.; Riese, D.J., 2nd; Hollenhorst, P.C.; Lorch, G.; Foley, A.M. At the crossroads: EGFR and PTHrP signaling in cancer-mediated diseases of bone. Odontology 2012, 100, 109–129. [Google Scholar] [CrossRef] [Green Version]
- Kir, S.; White, J.P.; Kleiner, S.; Kazak, L.; Cohen, P.; Baracos, V.E.; Spiegelman, B.M. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 2014, 513, 100–104. [Google Scholar] [CrossRef]
- Bing, C.; Brown, M.; King, P.; Collins, P.; Tisdale, M.J.; Williams, G. Increased gene expression of brown fat uncoupling protein (UCP)1 and skeletal muscle UCP2 and UCP3 in MAC16-induced cancer cachexia. Cancer Res. 2000, 60, 2405–2410. [Google Scholar]
- Brooks, S.L.; Neville, A.M.; Rothwell, N.J.; Stock, M.J.; Wilson, S. Sympathetic activation of brown-adipose-tissue thermogenesis in cachexia. Biosci. Rep. 1981, 1, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Shellock, F.G.; Riedinger, M.S.; Fishbein, M.C. Brown adipose tissue in cancer patients: Possible cause of cancer-induced cachexia. J. Cancer Res. Clin. Oncol. 1986, 111, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Tsoli, M.; Moore, M.; Burg, D.; Painter, A.; Taylor, R.; Lockie, S.H.; Turner, N.; Warren, A.; Cooney, G.; Oldfield, B.; et al. Activation of thermogenesis in brown adipose tissue and dysregulated lipid metabolism associated with cancer cachexia in mice. Cancer Res. 2012, 72, 4372–4382. [Google Scholar] [CrossRef]
- Kir, S.; Spiegelman, B.M. Cachexia & Brown Fat: A Burning Issue in Cancer. Trends Cancer 2016, 2, 461–463. [Google Scholar] [CrossRef]
- Song, W.; Kir, S.; Hong, S.; Hu, Y.; Wang, X.; Binari, R.; Tang, H.W.; Chung, V.; Banks, A.S.; Spiegelman, B.; et al. Tumor-Derived Ligands Trigger Tumor Growth and Host Wasting via Differential MEK Activation. Dev. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Yasmeen, R.; Shen, Q.; Lee, A.; Leung, J.H.; Kowdley, D.; DiSilvestro, D.J.; Xu, L.; Yang, K.; Maiseyeu, A.; Bal, N.C.; et al. Epiregulin induces leptin secretion and energy expenditure in high-fat diet-fed mice. J. Endocrinol. 2018, 239, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Hicks, M.J.; Chiuchiolo, M.J.; Ballon, D.; Dyke, J.P.; Aronowitz, E.; Funato, K.; Tabar, V.; Havlicek, D.; Fan, F.; Sondhi, D.; et al. Anti-Epidermal Growth Factor Receptor Gene Therapy for Glioblastoma. PLoS ONE 2016, 11, e0162978. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Udayakumar, D.; Cai, L.; Hu, Z.; Kapur, P.; Kho, E.Y.; Pavia-Jimenez, A.; Fulkerson, M.; de Leon, A.D.; Yuan, Q.; et al. Addressing metabolic heterogeneity in clear cell renal cell carcinoma with quantitative Dixon MRI. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, A.; Danai, L.V.; Vander Heiden, M.G. Microenvironmental regulation of cancer cell metabolism: Implications for experimental design and translational studies. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Hulea, L.; Gravel, S.P.; Morita, M.; Cargnello, M.; Uchenunu, O.; Im, Y.K.; Lehuede, C.; Ma, E.H.; Leibovitch, M.; McLaughlan, S.; et al. Translational and HIF-1alpha-Dependent Metabolic Reprogramming Underpin Metabolic Plasticity and Responses to Kinase Inhibitors and Biguanides. Cell Metab. 2018, 28, 817–832.e818. [Google Scholar] [CrossRef]
- Campbell, S.L.; Wellen, K.E. Metabolic Signaling to the Nucleus in Cancer. Mol. Cell 2018, 71, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic Instruction of Immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanford-Crane, H.; Abrego, J.; Sherman, M.H. Fibroblasts as Modulators of Local and Systemic Cancer Metabolism. Cancers 2019, 11, 619. https://doi.org/10.3390/cancers11050619
Sanford-Crane H, Abrego J, Sherman MH. Fibroblasts as Modulators of Local and Systemic Cancer Metabolism. Cancers. 2019; 11(5):619. https://doi.org/10.3390/cancers11050619
Chicago/Turabian StyleSanford-Crane, Hannah, Jaime Abrego, and Mara H. Sherman. 2019. "Fibroblasts as Modulators of Local and Systemic Cancer Metabolism" Cancers 11, no. 5: 619. https://doi.org/10.3390/cancers11050619
APA StyleSanford-Crane, H., Abrego, J., & Sherman, M. H. (2019). Fibroblasts as Modulators of Local and Systemic Cancer Metabolism. Cancers, 11(5), 619. https://doi.org/10.3390/cancers11050619