Insights into Telomerase/hTERT Alternative Splicing Regulation Using Bioinformatics and Network Analysis in Cancer


Abstract
:1. Introduction
2. Alternative Splicing is Dysregulated in Cancer Leading to the Re-Emergence of Splice Variants Normally Found in Development but Silenced in Normal Cells
3. Alternative Splicing of hTERT
3.1. hTERT Alternative Splicing during Human Embryogenesis and Development Indicates that Telomerase Activity is Regulated by Alternative Splicing
3.2. A Paradigm Shift: hTERT Is Regulated by Alternative Splicing in Cancers
3.3. RNA Sequencing and Other Technologies to Detect hTERT Splice Variants in Cancer
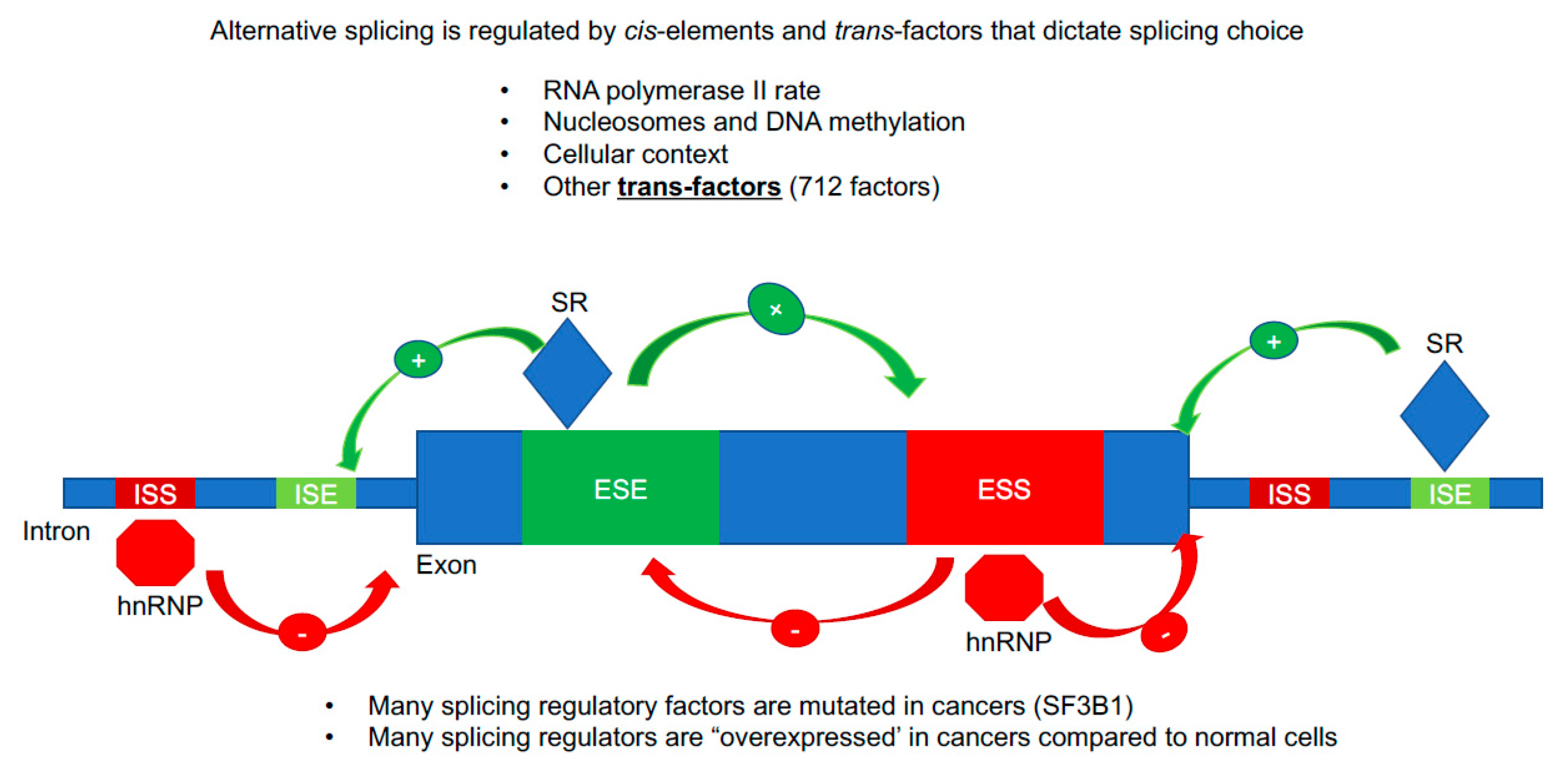
3.4. Regulation of hTERT Alternative Splicing by cis-Elements and trans-Factors
4. Using Bioinformatics to Discover hTERT Alternative Splicing Regulation in Cancers
5. Utilizing Predictive Models of RNA Folding and RNA trans-Factor Binding
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- De Lange, T. How shelterin solves the telomere end-protection problem. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 167–177. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Sfeir, A.; Gryaznov, S.M.; Shay, J.W.; Wright, W.E. Does a sentinel or a subset of short telomeres determine replicative senescence? Mol. Biol. Cell 2004, 15, 3709–3718. [Google Scholar] [CrossRef]
- Wright, W.E.; Shay, J.W. The two-stage mechanism controlling cellular senescence and immortalization. Exp. Gerontol. 1992, 27, 383–389. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, A.T.; Robin, J.D.; Sayed, M.; Litterst, C.M.; Shelton, D.N.; Shay, J.W.; Wright, W.E. Quantitative telomerase enzyme activity determination using droplet digital PCR with single cell resolution. Nucleic Acids Res. 2014, 42, e104. [Google Scholar] [CrossRef]
- Frink, R.E.; Peyton, M.; Schiller, J.H.; Gazdar, A.F.; Shay, J.W.; Minna, J.D. Telomerase inhibitor imetelstat has preclinical activity across the spectrum of non-small cell lung cancer oncogenotypes in a telomere length dependent manner. Oncotarget 2016, 7, 31639–31651. [Google Scholar] [CrossRef] [Green Version]
- Greider, C.W.; Blackburn, E.H. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 1987, 51, 887–898. [Google Scholar] [CrossRef]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Hiyama, K.; Hirai, Y.; Kyoizumi, S.; Akiyama, M.; Hiyama, E.; Piatyszek, M.A.; Shay, J.W.; Ishioka, S.; Yamakido, M. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J. Immunol. 1995, 155, 3711–3715. [Google Scholar] [PubMed]
- Avin, B.A.; Umbricht, C.B.; Zeiger, M.A. Human telomerase reverse transcriptase regulation by DNA methylation, transcription factor binding and alternative splicing (Review). Int. J. Oncol. 2016, 49, 2199–2205. [Google Scholar] [CrossRef] [PubMed]
- Leao, R.; Apolonio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: Clinical impacts in cancer. J. Biomed. Sci. 2018, 25, 22. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Chang, G.; Wang, F.; Wang, F.; Geng, X. Alternative Splicing of hTERT Pre-mRNA: A Potential Strategy for the Regulation of Telomerase Activity. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.; Wright, W.E.; Shay, J.W. Alternative splicing regulation of telomerase: A new paradigm? Trends Genet. 2014, 30, 430–438. [Google Scholar] [CrossRef]
- Kim, W.; Ludlow, A.T.; Min, J.; Robin, J.D.; Stadler, G.; Mender, I.; Lai, T.P.; Zhang, N.; Wright, W.E.; Shay, J.W. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect-Over Long Distances (TPE-OLD): Implications for Aging and Cancer. PLoS Biol. 2016, 14, e2000016. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, A.T.; Wong, M.S.; Robin, J.D.; Batten, K.; Yuan, L.; Lai, T.-P.; Dahlson, N.; Zhang, L.; Mender, I.; Tedone, E.; et al. NOVA1 regulates hTERT splicing and cell growth in non-small cell lung cancer. Nat. Commun. 2018, 9, 3112. [Google Scholar] [CrossRef]
- Sayed, M.E.; Yuan, L.; Robin, J.D.; Tedone, E.; Batten, K.; Dahlson, N.; Wright, W.E.; Shay, J.W.; Ludlow, A.T. NOVA1 directs PTBP1 to hTERT pre-mRNA and promotes telomerase activity in cancer cells. Oncogene 2019, 38, 2937–2952. [Google Scholar] [CrossRef]
- Withers, J.B.; Ashvetiya, T.; Beemon, K.L. Exclusion of exon 2 is a common mRNA splice variant of primate telomerase reverse transcriptases. PLoS ONE 2012, 7, e48016. [Google Scholar] [CrossRef]
- Yi, X.; Shay, J.W.; Wright, W.E. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res. 2001, 29, 4818–4825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, X.; Tesmer, V.M.; Savre-Train, I.; Shay, J.W.; Wright, W.E. Both transcriptional and posttranscriptional mechanisms regulate human telomerase template RNA levels. Mol. Cell. Biol. 1999, 19, 3989–3997. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W. Telomerase RNA levels limit the telomere length equilibrium. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 225–229. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef]
- Akincilar, S.C.; Low, K.C.; Liu, C.Y.; Yan, T.D.; Oji, A.; Ikawa, M.; Li, S.; Tergaonkar, V. Quantitative assessment of telomerase components in cancer cell lines. FEBS Lett. 2015, 589, 974–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, L.; Cech, T.R. Inventory of telomerase components in human cells reveals multiple subpopulations of hTR and hTERT. Nucleic Acids Res. 2014, 42, 8565–8577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burchett, K.M.; Yan, Y.; Ouellette, M.M. Telomerase inhibitor Imetelstat (GRN163L) limits the lifespan of human pancreatic cancer cells. PLoS ONE 2014, 9, e85155. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef]
- Marian, C.O.; Cho, S.K.; McEllin, B.M.; Maher, E.A.; Hatanpaa, K.J.; Madden, C.J.; Mickey, B.E.; Wright, W.E.; Shay, J.W.; Bachoo, R.M. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin. Cancer Res. 2010, 16, 154–163. [Google Scholar] [CrossRef]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Holohan, B.; Hagiopian, M.M.; Lai, T.P.; Huang, E.; Friedman, D.R.; Wright, W.E.; Shay, J.W. Perifosine as a potential novel anti-telomerase therapy. Oncotarget 2015, 6, 21816–21826. [Google Scholar] [CrossRef] [Green Version]
- Buseman, C.M.; Wright, W.E.; Shay, J.W. Is telomerase a viable target in cancer? Mutat. Res. 2012, 730, 90–97. [Google Scholar] [CrossRef]
- Reyes-Uribe, P.; Adrianzen-Ruesta, M.P.; Deng, Z.; Echevarria-Vargas, I.; Mender, I.; Saheb, S.; Liu, Q.; Altieri, D.C.; Murphy, M.E.; Shay, J.W.; et al. Exploiting TERT dependency as a therapeutic strategy for NRAS-mutant melanoma. Oncogene 2018, 37, 4058–4072. [Google Scholar] [CrossRef] [PubMed]
- Mender, I.; Gryaznov, S.; Dikmen, Z.G.; Wright, W.E.; Shay, J.W. Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2′-deoxyguanosine. Cancer Discov. 2015, 5, 82–95. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Black, D.L. Protein diversity from alternative splicing: A challenge for bioinformatics and post-genome biology. Cell 2000, 103, 367–370. [Google Scholar] [CrossRef]
- Taft, R.J.; Pheasant, M.; Mattick, J.S. The relationship between non-protein-coding DNA and eukaryotic complexity. Bioessays 2007, 29, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Hillman, R.T.; Green, R.E.; Brenner, S.E. An unappreciated role for RNA surveillance. Genome Biol. 2004, 5, R8. [Google Scholar] [CrossRef]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schor, I.E.; Gomez Acuna, L.I.; Kornblihtt, A.R. Coupling between transcription and alternative splicing. Cancer Treat. Res. 2013, 158, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Nojima, T.; Rebelo, K.; Gomes, T.; Grosso, A.R.; Proudfoot, N.J.; Carmo-Fonseca, M. RNA Polymerase II Phosphorylated on CTD Serine 5 Interacts with the Spliceosome during Co-transcriptional Splicing. Mol. Cell 2018, 72, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Watson, M. The incredible complexity of RNA splicing. Genome Biol. 2016, 17, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, D.; Freese, P.; Alexis, M.S.; Su, A.; Hochman, M.; Palden, T.; Bazile, C.; Lambert, N.J.; Van Nostrand, E.L.; Pratt, G.A.; et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 2018, 70, 854–867. [Google Scholar] [CrossRef]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Nehyba, J.; Bose, H.R., Jr. Alternatively spliced telomerase reverse transcriptase variants lacking telomerase activity stimulate cell proliferation. Mol. Cell. Biol. 2012, 32, 4283–4296. [Google Scholar] [CrossRef]
- Listerman, I.; Sun, J.; Gazzaniga, F.S.; Lukas, J.L.; Blackburn, E.H. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res. 2013, 73, 2817–2828. [Google Scholar] [CrossRef]
- Yi, X.; White, D.M.; Aisner, D.L.; Baur, J.A.; Wright, W.E.; Shay, J.W. An alternate splicing variant of the human telomerase catalytic subunit inhibits telomerase activity. Neoplasia 2000, 2, 433–440. [Google Scholar] [CrossRef]
- Fleisig, H.B.; Hukezalie, K.R.; Thompson, C.A.; Au-Yeung, T.T.; Ludlow, A.T.; Zhao, C.R.; Wong, J.M. Telomerase reverse transcriptase expression protects transformed human cells against DNA-damaging agents, and increases tolerance to chromosomal instability. Oncogene 2016, 35, 218–227. [Google Scholar] [CrossRef]
- Zhu, S.; Rousseau, P.; Lauzon, C.; Gandin, V.; Topisirovic, I.; Autexier, C. Inactive C-terminal telomerase reverse transcriptase insertion splicing variants are dominant-negative inhibitors of telomerase. Biochimie 2014, 101, 93–103. [Google Scholar] [CrossRef]
- Ulaner, G.A.; Hu, J.F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. 1998, 58, 4168–4172. [Google Scholar]
- Teichroeb, J.H.; Kim, J.; Betts, D.H. The role of telomeres and telomerase reverse transcriptase isoforms in pluripotency induction and maintenance. RNA Biol. 2016, 13, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.D.; Brenner, S.E.; Dudoit, S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010, 38, e131. [Google Scholar] [CrossRef]
- Bayega, A.; Wang, Y.C.; Oikonomopoulos, S.; Djambazian, H.; Fahiminiya, S.; Ragoussis, J. Transcript Profiling Using Long-Read Sequencing Technologies. Methods Mol. Biol. 2018, 1783, 121–147. [Google Scholar] [CrossRef]
- Wong, M.S.; Chen, L.; Foster, C.; Kainthla, R.; Shay, J.W.; Wright, W.E. Regulation of telomerase alternative splicing: A target for chemotherapy. Cell Rep. 2013, 3, 1028–1035. [Google Scholar] [CrossRef]
- Wong, M.S.; Shay, J.W.; Wright, W.E. Regulation of human telomerase splicing by RNA:RNA pairing. Nat. Commun. 2014, 5, 3306. [Google Scholar] [CrossRef] [Green Version]
- Bojesen, S.E.; Pooley, K.A.; Johnatty, S.E.; Beesley, J.; Michailidou, K.; Tyrer, J.P.; Edwards, S.L.; Pickett, H.A.; Shen, H.C.; Smart, C.E.; et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet. 2013, 45, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Killedar, A.; Stutz, M.D.; Sobinoff, A.P.; Tomlinson, C.G.; Bryan, T.M.; Beesley, J.; Chenevix-Trench, G.; Reddel, R.R.; Pickett, H.A. A Common Cancer Risk-Associated Allele in the hTERT Locus Encodes a Dominant Negative Inhibitor of Telomerase. PLoS Genet. 2015, 11, e1005286. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khattar, E.; Kumar, P.; Liu, C.Y.; Akincilar, S.C.; Raju, A.; Lakshmanan, M.; Maury, J.J.; Qiang, Y.; Li, S.; Tan, E.Y.; et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J. Clin. Invest. 2016, 126, 4045–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Cheng, Y.; Zhang, C.; Chang, G.; Geng, X. A novel antisense oligonucleotide anchored on the intronic splicing enhancer of hTERT pre-mRNA inhibits telomerase activity and induces apoptosis in glioma cells. J. Neurooncol. 2019, 143, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Fallmann, J.; Will, S.; Engelhardt, J.; Gruning, B.; Backofen, R.; Stadler, P.F. Recent advances in RNA folding. J. Biotechnol. 2017, 261, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Cui, J.; Cheng, J.; Wu, R. Computational Prediction of RNA-Binding Proteins and Binding Sites. Int. J. Mol. Sci. 2015, 16, 26303–26317. [Google Scholar] [CrossRef] [Green Version]
- Buckanovich, R.J.; Posner, J.B.; Darnell, R.B. Nova, the paraneoplastic Ri antigen, is homologous to an RNA-binding protein and is specifically expressed in the developing motor system. Neuron 1993, 11, 657–672. [Google Scholar] [CrossRef]
- Jensen, K.B.; Dredge, B.K.; Stefani, G.; Zhong, R.; Buckanovich, R.J.; Okano, H.J.; Yang, Y.Y.; Darnell, R.B. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 2000, 25, 359–371. [Google Scholar] [CrossRef]
- Saito, Y.; Miranda-Rottmann, S.; Ruggiu, M.; Park, C.Y.; Fak, J.J.; Zhong, R.; Duncan, J.S.; Fabella, B.A.; Junge, H.J.; Chen, Z.; et al. NOVA2-mediated RNA regulation is required for axonal pathfinding during development. eLife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Dredge, B.K.; Stefani, G.; Engelhard, C.C.; Darnell, R.B. Nova autoregulation reveals dual functions in neuronal splicing. EMBO J. 2005, 24, 1608–1620. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isoform | Exon Structure | Intron Retention? | Biochemical Function |
|---|---|---|---|
| Full-length | 1–16. Original ORF. | No | Functional hTERT protein, maintains telomeres when in active telomerase holoenzyme (RNP) |
| Minus beta | 1–6, 9, and 10; PTC in 10. Skipping of exons 7 and 8. | No | Mostly degraded by non-sense mediated decay, some translated into protein and may play a role in DNA damage repair/ protection from apoptosis, may bind hTERC (hTR) |
| Minus alpha | 1–16, alternative 3′ splice acceptor site in exon 6 generates in frame shift of 36 nucleotides. Original ORF. | No | Dominant-negative, binds hTERC (hTR) |
| INS3 | 1–16 plus, PTC in intron 14. | Retention of intron 14 nucleotide 623 to end of intron 14. | Dominant-negative, binds hTERC (hTR) |
| INS4 | 1–14, and alternative exon 16 3′ splice site NT492, PTC in exon 14. | Retention of intron 14 nucleotides 1–600. | Dominant-negative, binds hTERC (hTR) |
| DEL2 | 1,3–16, PTC in exon 3. | No | Proposed mitochondrial hTERT variant, retains hTERT MLS in exon 1. |
| Delta4–13 | 1–3, 14–16, original ORF. | No | Proposed to stimulate proliferation. Interacts with WNT/Beta catenin. |
| Minus Gamma | Skipping of exon 11. Original ORF. | No | Tissue specific and may inhibit telomerase action at the telomeres. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ludlow, A.T.; Slusher, A.L.; Sayed, M.E. Insights into Telomerase/hTERT Alternative Splicing Regulation Using Bioinformatics and Network Analysis in Cancer. Cancers 2019, 11, 666. https://doi.org/10.3390/cancers11050666
Ludlow AT, Slusher AL, Sayed ME. Insights into Telomerase/hTERT Alternative Splicing Regulation Using Bioinformatics and Network Analysis in Cancer. Cancers. 2019; 11(5):666. https://doi.org/10.3390/cancers11050666
Chicago/Turabian StyleLudlow, Andrew T., Aaron L. Slusher, and Mohammed E. Sayed. 2019. "Insights into Telomerase/hTERT Alternative Splicing Regulation Using Bioinformatics and Network Analysis in Cancer" Cancers 11, no. 5: 666. https://doi.org/10.3390/cancers11050666
APA StyleLudlow, A. T., Slusher, A. L., & Sayed, M. E. (2019). Insights into Telomerase/hTERT Alternative Splicing Regulation Using Bioinformatics and Network Analysis in Cancer. Cancers, 11(5), 666. https://doi.org/10.3390/cancers11050666