Malignant Pheochromocytomas/Paragangliomas and Ectopic Hormonal Secretion: A Case Series and Review of the Literature



 , ,
, , 

Abstract
:1. Introduction
2. Results
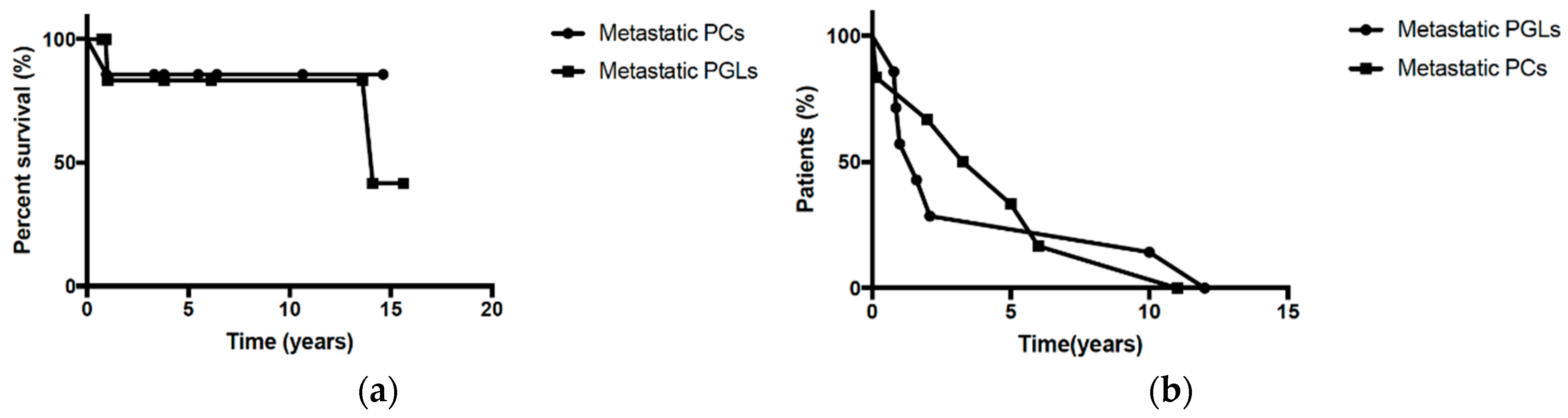
2.1. Epidemiological and Clinicopathological Data
2.2. Treatment and Outcome
2.3. Ectopic Secretion and Review of the Literature
3. Discussion
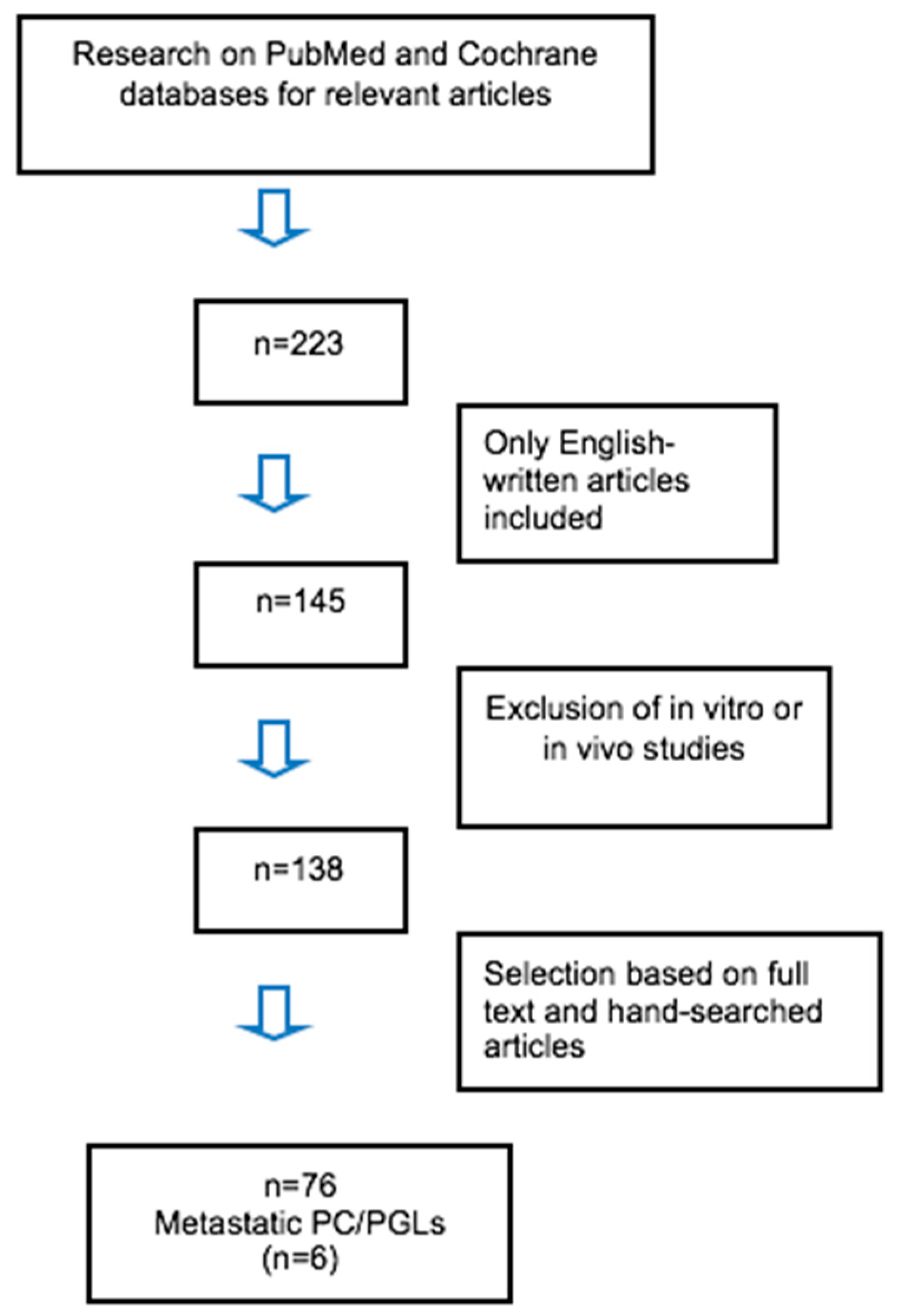
4. Methods
4.1. Patients
4.2. Review of the Literature on Ectopic Hormonal Secretion
4.3. Hormonal Secretion
4.4. Imaging
4.5. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lenders, J.; Eisenhofer, G.; Mannelli, M.; Pacak, K. Phaeochromocytoma. Lancet 2005, 366, 665–675. [Google Scholar] [CrossRef]
- Chrisoulidou, A.; Kaltsas, G.; Ilias, I.; Grossman, A.B. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr. Relat. Cancer 2007, 14, 569–585. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IRAC). WHO Classification of Tumors of Endocrine Organs; World Health Organization: Lyon, France, 2017. [Google Scholar]
- Tischler, A.S.; de Krijger, R.R. Phaeochromocytoma. In WHO Classification of Tumors of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloppel, G., Eds.; IARC Press: Lyons, France, 2017; pp. 183–189. [Google Scholar]
- Ayala-Ramirez, M.; Feng, L.; Johnson, M.M.; Ejaz, S.; Habra, M.A.; Rich, T.; Busaidy, N.; Cote, G.J.; Perrier, N.; Phan, A.; et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: Primary tumor size and primary tumor location as prognostic indicators. J. Clin. Endocrinol. Metab. 2011, 96, 717–725. [Google Scholar] [CrossRef]
- Huang, K.H.; Chung, S.D.; Chen, S.C.; Chueh, S.C.; Pu, Y.S.; Lai, M.K.; Lin, W.C. Clinical and pathological data of 10 malignant pheochromocytomas: Long-term follow up in a single institute. Int. J. Urol. 2007, 14, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Prejbisz, A.; Lenders, J.W.; Eisenhofer, G.; Januszewicz, A. Mortality associated with phaeochromocytoma. Horm. Metab. Res. 2013, 45, 154–158. [Google Scholar] [CrossRef]
- Amar, L.; Lussey-Lepoutre, C.; Lenders, J.W.; Djadi-Prat, J.; Plouin, P.F.; Steichen, O. Management of endocrine disease: Recurrence or new tumors after complete resection of pheochromocytomas and paragangliomas: A systematic review and meta-analysis. Eur. J. Endocrinol. 2016, 175, 135–145. [Google Scholar] [CrossRef]
- Bravo, E.L.; Tagle, R. Pheochromocytoma: State-of-the-art and future prospects. Endocr. Rev. 2003, 24, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Takekoshi, K.; Naruse, M. Risk Stratification on Pheochromocytoma and Paraganglioma from Laboratory and Clinical Medicine. J. Clin. Med. 2018, 7, 242. [Google Scholar] [CrossRef]
- Mlika, M.; Kourda, N.; Zorgati, M.M.; Bahri, S.; Ben Ammar, S.; Zermani, R. Prognostic value of Pheochromocytoma of the Adrenal Gland Scaled Score (Pass score) tests to separate benign from malignant neoplasms. Tunis Med. 2013, 91, 209–215. [Google Scholar] [PubMed]
- Kim, K.Y.; Kim, J.H.; Hong, A.R.; Seong, M.W.; Lee, K.E.; Kim, S.J.; Kim, S.W.; Shin, C.S.; Kim, S.Y. Disentangling of malignancy from benign pheochromocytomas/paragangliomas. PLoS ONE 2016, 11, e0168413. [Google Scholar] [CrossRef] [PubMed]
- Zelinka, T.; Musil, Z.; Dušková, J.; Burton, D.; Merino, M.J.; Milosevic, D.; Widimský, J.; Pacak, K. Metastatic pheochromocytoma: Does the size and age matter? Eur. J. Clin. Investig. 2011, 41, 1121–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidi, O.; Young, W.F.; Iñiguez-Ariza, N.M.; Kittah, N.E.; Gruber, L.; Bancos, C.; Tamhane, S.; Bancos, I. Malignant Pheochromocytoma and Paraganglioma: 272 Patients Over 55 Years. J. Clin. Endocrinol. Metab. 2017, 102, 3296–3305. [Google Scholar] [CrossRef] [PubMed]
- Van der Harst, E.; de Herder, W.W.; de Krijger, R.R.; Bruining, H.A.; Bonjer, H.J.; Lamberts, S.W.; van den Meiracker, A.H.; Stijnen, T.H.; Boomsma, F. The value of plasma markers for the clinical behaviour of phaeochromocytomas. Eur. J. Endocrinol. 2002, 147, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Plouin, P.F.; Chatellier, G.; Grouzmann, E.; Azizi, M.; Denolle, T.; Comoy, E.; Corvol, P. Plasma neuropeptide Y and catecholamine concentrations and urinary metanephrine excretion in patients with adrenal or ectopic phaeochromocytoma. J. Hypertens. 1991, 9, 272–273. [Google Scholar]
- Toledo, R.; Jimenez, C. Recent advances in the management of malignant pheochromocytoma and paraganglioma: Focus on tyrosine kinase and hypoxia-inducible factor inhibitors. F1000Res. 2018, 30, 7. [Google Scholar] [CrossRef]
- Hescot, S.; Leboulleux, S.; Amar, L.; Vezzosi, D.; Borget, I.; Bournaud-Salinas, C.; de la Fouchardiere, C.; Libé, R.; Do Cao, C.; Niccoli, P.; et al. French Group of Endocrine and Adrenal Tumors (Groupe des Tumeurs Endocrines-REseau NAtional des Tumeurs ENdocrines and COrtico-MEdullo Tumeurs Endocrines Networks Neuroendocrine Tumors. NCCN Guidelines. 2017. Available online: http://www.NCCN.org (accessed on 2 May 2019).
- Mak, I.Y.F.; Hayes, A.R.; Khoo, B.; Grossman, A. Peptide Receptor Radionuclide Therapy as a Novel Treatment for Metastatic and Invasive Phaeochromocytoma and Paraganglioma. Neuroendocrinology 2019, 12. [Google Scholar] [CrossRef]
- McBride, J.F.; Atwell, T.D.; Charboneau, W.J.; Young, W.F.; Wass, T.C.; Callstrom, M.R. Minimally invasive treatment of metastatic pheochromocytoma and paraganglioma: Efficacy and safety of radiofrequency ablation and cryoablation therapy. J. Vasc. Interv. Radiol. 2011, 22, 1263–1270. [Google Scholar] [CrossRef]
- Ballav, C.; Naziat, A.; Mihai, R.; Karavitaki, N.; Ansorge, O.; Grossman, A.B. Mini-review: Pheochromocytomas causing the ectopic ACTH syndrome. Endocrine 2012, 42, 69–73. [Google Scholar] [CrossRef]
- Lois, K.B.; Santhakumar, A.; Vaikkakara, S.; Mathew, S.; Long, A.; Johnson, S.J.; Peaston, R.; Neely, R.D.G.; Richardson, D.L.; Graham, J.; et al. Phaeochromocytoma and ACTH-dependent cushing’s syndrome: Tumor secretion can mimic pituitary cushing’s disease. Clin. Endocrinol. (Oxf.) 2016, 84, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Araujo Castro, M.; Palacios García, N.; Aller Pardo, J.; Izquierdo Alvarez, C.; Armengod Grao, L.; Estrada García, J. Ectopic Cushing syndrome: Report of 9 cases. Endocrinol. Diabetes Nutr. 2018, 65, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, I.; Higuchi, S.; Fujimoto, M.; Takiguchi, T.; Nakayama, A.; Tamura, A.; Kohno, T.; Komai, E.; Shiga, A.; Nagano, H.; et al. Cushing Sndrome Due to ACTH-Secreting Pheochromocytoma, Aggravated by Glucocorticoid-Driven Positive-Feedback Loop. J. Clin. Endocrinol. Metab. 2016, 101, 841–846. [Google Scholar] [CrossRef]
- Kakudo, K.; Uematsu, K.; Matsuno, Y.; Mitsunobu, M.; Toyosaka, A.; Okamoto, E.; Fukuchi, M. Malignant pheochromocytoma with ACTH production. Acta Pathol. Jpn. 1984, 34, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Brenner, N.; Kopetschke, R.; Ventz, M.; Strasburger, C.J.; Quinkler, M.; Gerl, H. Cushing’s syndrome due to ACTH-secreting pheochromocytoma. Can. J. Urol. 2008, 15, 3924–3927. [Google Scholar]
- Otsuka, F.; Miyoshi, T.; Murakami, K.; Inagaki, K.; Takeda, M.; Ujike, K.; Ogura, T.; Omori, M.; Doihara, H.; Tanaka, Y.; et al. An extra-adrenal abdominal pheochromocytoma causing ectopic ACTH syndrome. Am. J. Hypertens. 2005, 18, 1364–1368. [Google Scholar] [CrossRef]
- Beaser, R.S.; Guay, A.T.; Lee, A.K.; Silverman, M.L.; Flint, L.D. An adrenocorticotropic hormone-producing pheochromocytoma: Diagnostic and immunohistochemical studies. J. Urol. 1986, 135, 10–13. [Google Scholar] [CrossRef]
- Chen, H.; Doppman, J.L.; Chrousos, G.P.; Norton, J.A.; Nieman, L.K.; Udelsman, R. Adrenocorticotropic hormone-secreting pheochromocytomas: The exception to the rule. Surgery 1995, 118, 988–994. [Google Scholar] [CrossRef]
- Tutal, E.; Yılmazer, D.; Demirci, T.; Cakır, E.; Gültekin, S.S.; Celep, B.; Topaloğlu, O.; Çakal, E. A rare case of ectopic ACTH syndrome originating from malignant renal paraganglioma. Arch. Endocrinol. Metab. 2017, 61, 291–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apple, D.; Kreines, K. Cushing’s syndrome due to ectopic ACTH production by a nasal paraganglioma. Am. J. Med. Sci. 1982, 283, 32–35. [Google Scholar] [CrossRef]
- Dahir, K.M.; Gonzalez, A.; Revelo, M.P.; Ahmed, S.R.; Roberts, J.R.; Blevins, L.S., Jr. Ectopic adrenocorticotropic hormone hypersecretion due to a primary pulmonary paraganglioma. Endocr. Pract. 2004, 10, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Kirkby-Bott, J.; Brunaud, L.; Mathonet, M.; Hamoir, E.; Kraimps, J.L.; Trésallet, C.; Amar, L.; Rault, A.; Henry, J.F.; Carnaille, B. Ectopic hormone-secreting pheochromocytoma: A francophone observational study. World J. Surg. 2012, 36, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, P.; Isidro, L.; González-Martín, M.; Loidi, L.; Arnal, F.; Cordido, F. Ectopic adrenocorticotropic hormone production by a noncatecholamine secreting pheochromocytoma. J. Urol. 2002, 167, 2514–2515. [Google Scholar] [CrossRef]
- White, A.; Ray, D.W.; Talbot, A.; Abraham, P.; Thody, A.J.; Bevan, J.S. Cushing’s syndrome due to phaeochromocytoma secreting the precursors of adrenocorticotropin. J. Clin. Endocrinol. Metab. 2000, 85, 4771–4775. [Google Scholar] [CrossRef]
- Loh, K.C.; Gupta, R.; Shlossberg, A.H. Spontaneous remission of ectopic Cushing’s syndrome due to pheochromocytoma: A case report. Eur. J. Endocrinol. 1996, 135, 440–443. [Google Scholar] [CrossRef]
- Terzolo, M.; Alì, A.; Pia, A.; Bollito, E.; Reimondo, G.; Paccotti, P.; Scardapane, R.; Angeli, A. Cyclic Cushing’s syndrome due to ectopic ACTH secretion by an adrenal pheochromocytoma. J. Endocrinol. Investig. 1994, 17, 869–874. [Google Scholar] [CrossRef]
- Mendonça, B.B.; Arnhold, I.J.; Nicolau, W.; Avancini, V.A.; Boise, W. Cushing’s syndrome due to ectopic ACTH secretion by bilateral pheochromocytomas in multiple endocrine neoplasia type 2A. N. Engl. J. Med. 1988, 319, 1610–1611. [Google Scholar]
- Schroeder, J.O.; Asa, S.L.; Kovacs, K.; Killinger, D.; Hadley, G.L.; Volpé, R. Report of a case of pheochromocytoma producing immunoreactive ACTH and beta-endorphin. J. Endocrinol. Investig. 1984, 7, 117–121. [Google Scholar] [CrossRef]
- Van Brummelen, P.; Van Hooff, J.P.; Van Seters, A.P.; Giard, R.W. Ectopic ACTH production by a functioning phaeochromocytoma. Neth. J. Med. 1982, 25, 237–241. [Google Scholar]
- Spark, R.F.; Connolly, P.B.; Gluckin, D.S.; White, R.; Sacks, B.; Landsberg, L. ACTH secretion from a functioning pheochromocytoma. N. Engl. J. Med. 1979, 301, 416–418. [Google Scholar] [CrossRef]
- Thomas, T.; Zender, S.; Terkamp, C.; Jaeckel, E.; Manns, M.P. Hypercortisolaemia due to ectopic adrenocorticotropic hormone secretion by a nasal paraganglioma: A case report and review of the literature. BMC Res. Notes 2013, 19, 331. [Google Scholar] [CrossRef]
- Serra, F.; Duarte, S.; Abreu, S.; Marques, C.; Cassis, J.; Saraiva, M. Cushing’s syndrome due to ectopic ACTH production by a nasal paraganglioma. Endocrinol. Diabetes Metab. Case Rep. 2013, 2013, 130038. [Google Scholar] [CrossRef]
- Folkestad, L.; Andersen, M.S.; Nielsen, A.L.; Glintborg, D. A rare cause of Cushing’s syndrome: An ACTH-secreting phaeochromocytoma. BMJ Case Rep. 2014, 8, 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, X.G.; Zhang, D.X.; Li, X.; Cui, X.G.; Xu, D.F.; Li, Y.; Gao, Y.; Yin, L.; Ren, J.Z. Adrenocorticotropic hormone-producing pheochromocytoma: A case report and review of the literature. Chin. Med. J. 2012, 125, 1193–1196. [Google Scholar]
- Bernardi, S.; Grimaldi, F.; Finato, N.; De Marchi, S.; Proclemer, A.; Sabato, N.; Bertolotto, M.; Fabris, B. A pheochromocytoma with high adrenocorticotropic hormone and a silent lung nodule. Am. J. Med. Sci. 2011, 342, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Nijhoff, M.F.; Dekkers, O.M.; Vleming, L.J.; Smit, J.W.; Romijn, J.A.; Pereira, A.M. ACTH-producing pheochromocytoma: Clinical considerations and concise review of the literature. Eur. J. Intern. Med. 2009, 20, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, P.S.; van Gils, A.; Canninga-van Dijk, M.R.; de Koning, E.J.; Hofland, L.J.; de Herder, W.W. Sequential ACTH and catecholamine secretion in a phaeochromocytoma. Eur. J. Endocrinol. 2002, 147, 201–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberum, B.; Jaspers, C.; Munzenmaier, R. ACTH-producing paraganglioma of the paranasal sinuses. HNO 2003, 51, 328–331. [Google Scholar] [CrossRef]
- Forman, B.H.; Marban, E.; Kayne, R.D.; Passarelli, N.M.; Bobrow, S.N.; Livolsi, V.A.; Merino, M.; Minor, M.; Farber, L.R. Ectopic ACTH syndrome due to pheochromocytoma: Case report and review of the literature. Yale J. Biol. Med. 1979, 52, 181–189. [Google Scholar]
- Inoue, M.; Okamura, K.; Kitaoka, C.; Kinoshita, F.; Namitome, R.; Nakamura, U.; Shiota, M.; Goto, K.; Ohtsubo, T.; Matsumura, K.; et al. Metyrapone-responsive ectopic ACTH-secreting pheochromocytoma with a vicious cycle via a glucocorticoid-driven positive-feedback mechanism. Endocr. J. 2018, 65, 755–767. [Google Scholar] [CrossRef]
- Eng, P.H.K.; Tan, L.H.C.; Wong, K.S.; Cheng, C.W.; Fok, A.C.K.; Khoo, D.H.C. Cushing’s syndrome in a patient with a corticotropin-releasing hormone-producing pheochromocytoma. Endocr. Pr. 1999, 5, 84–87. [Google Scholar]
- Ruggeri, R.M.; Ferraù, F.; Campennì, A.; Simone, A.; Barresi, V.; Giuffrè, G.; Tuccari, G.; Baldari, S.T.F. Immunohistochemical localization and functional characterization of somatostatin receptor subtypes in a corticotropin releasing hormone- secreting adrenal phaeochromocytoma: Review of the literature and report of a case. Eur. Histochem. 2009, 53, 1–6. [Google Scholar] [CrossRef]
- Bayraktar, F.; Kebapcilar, L.; Kocdor, M.A.; Asa, S.L.; Yesil, S.; Canda, S.; Demir, T.; Saklamaz, A.; Seçil, M.; Akinci, B.; et al. Cushing’s syndrome due to ectopic CRH secretion by adrenal pheochromocytoma accompanied by renal infarction. Exp. Clin. Endocrinol. Diabetes 2006, 114, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Jessop, D.S.; Cunnah, D.; Millar, J.G.; Neville, E.; Coates, P.; Doniach, I.; Besser, G.M.R.L. A phaeochromocytoma presenting with Cushing’s syndrome associated with increased concentrations of circulating corticotrophin-releasing factor. J. Endocrinol. 1987, 113, 133–138. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.; Young, W.F., Jr.; Davila, D.G.; Scheithauer, B.W.; Kovacs, K.; Horvath, E.; Vale, W.; van Heerden, J.A. Cushing’s syndrome associated with ectopic production of corticotrophin-releasing hormone, corticotrophin and vasopressin by a phaeochromocytoma. Clin. Endocrinol. 1992, 37, 460–467. [Google Scholar] [CrossRef]
- Iwayama, H.; Hirase, S.; Nomura, Y.; Ito, T.; Morita, H.; Otake, K.; Okumura, A.; Takagi, J. Spontaneous adrenocorticotropic hormone (ACTH) normalisation due to tumor regression induced by metyrapone in a patient with ectopic ACTH syndrome: Case report and literature review. BMC Endocr. Disord. 2018, 18, 19. [Google Scholar] [CrossRef] [PubMed]
- Willenberg, H.S.; Feldkamp, J.; Lehmann, R.; Schott, M.; Goretzki, P.E.; Scherbaum, W.A. A case of catecholamine and glucocorticoid excess syndrome due to a corticotropin-secreting paraganglioma. Ann. N. Y. Acad. Sci. 2006, 1073, 52. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Suemaru, S.; Hattori, T.; Sugawara, M.; Ota, Z.; Takata, S.; Hamaya, K.; Doi, K.; Chrétien, M. Multiple endocrine neoplasia with Cushing’s syndrome due to paraganglioma producing corticotropin-releasing factor and adrenocorticotropin. Acta Endocrinol. (Cph.) 1986, 113, 189–195. [Google Scholar] [CrossRef]
- Sackel, S.G.; Manson, J.E.; Harawi, S.J.; Burakoff, R. Watery diarrhea syndrome due to an adrenal pheochromocytoma secreting vasoactive intestinal polypeptide. Dig. Dis. Sci. 1985, 30, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Cooperman, A.M.; Desantis, D.; Winkelman, E.; Farmer, R.; Eversman, J.; Said, S. Watery diarrhea syndrome. Two unusual cases and further evidence that VIP is a humoral mediator. Ann. Surg. 1978, 187, 325–328. [Google Scholar] [CrossRef]
- Herrera, M.F.; Stone, E.; Deitel, M.; Asa, S.L. Pheochromocytoma producing multiple vasoactive peptides. Arch. Surg. 1992, 127, 105–108. [Google Scholar] [CrossRef]
- Stewart, A.F.; Hoecker, J.L.; Mallette, L.E.; Segre, G.V.; Amatruda, T.T., Jr.; Vignery, A. Hypercalcemia in pheochromocytoma. Evidence for a novel mechanism. Ann. Intern. Med. 1985, 102, 776–779. [Google Scholar] [CrossRef]
- Suzuki, K.; Miyashita, A.; Inoue, Y.; Iki, S.; Enomoto, H.; Takahashi, Y.; Takemura, T. Interleukin-6-producing pheochromocytoma. Acta Haematol. 1991, 85, 217–219. [Google Scholar] [CrossRef]
- Takagi, M.; Egawa, T.; Motomura, T.; Sakuma-Mochizuki, J.; Nishimoto, N.; Kasayama, S.; Hayashi, S.; Koga, M.; Yoshizaki, K.; Yoshioka, T.; et al. Interleukin-6 secreting phaeochromocytoma associated with clinical markers of inflammation. Clin. Endocrinol. (Oxf.) 1997, 46, 507–509. [Google Scholar] [CrossRef]
- Shimizu, C.; Kubo, M.; Takano, K.; Takano, A.; Kijima, H.; Saji, H.; Katsuyama, I.; Sasano, H.; Koike, T. Interleukin-6 (IL-6) producing phaeochromocytoma: Direct IL-6 suppression by non-steroidal anti-inflammatory drugs. Clin. Endocrinol. (Oxf.) 2001, 54, 405–410. [Google Scholar] [CrossRef]
- Tokuda, H.; Hosoi, T.; Hayasaka, K.; Okamura, K.; Yoshimi, N.; Kozawa, O. Overexpression of protein kinase C-delta plays a crucial role in interleukin-6-producing pheochromocytoma presenting with acute inflammatory syndrome: A case report. Horm. Metab. Res. 2009, 41, 333–338. [Google Scholar] [CrossRef]
- Fukumoto, S.; Matsumoto, T.; Harada, S.; Fujisaki, J.; Kawano, M.; Ogata, E. Pheochromocytoma with pyrexia and marked inflammatory signs: A paraneoplastic syndrome with possible relation to interleukin-6 production. J. Clin. Endocrinol. Metab. 1991, 73, 877–881. [Google Scholar] [CrossRef]
- Minetto, M.; Dovio, A.; Ventura, M.; Cappia, S.; Daffara, F.; Terzolo, M.; Angeli, A. Interleukin-6 producing pheochromocytoma presenting with acute inflammatory syndrome. J. Endocrinol. Investig. 2003, 26, 453–457. [Google Scholar] [CrossRef]
- Kang, J.M.; Lee, W.J.; Kim, W.B.; Kim, T.Y.; Koh, J.M.; Hong, S.J.; Huh, J.R.J.Y.; Chi, H.S.; Kim, M.S. Systemic inflammatory syndrome and hepatic inflammatory cell infiltration caused by an interleukin-6 producing pheochromocytoma. Endocr. J. 2005, 52, 193–198. [Google Scholar] [CrossRef]
- Salahuddin, A.; Rohr-Kirchgraber, T.; Shekar, R.; West, B.; Loewenstein, J. Interleukin-6 in the fever and multiorgan crisis of pheochromocytoma. Scand. J. Infect. Dis. 1997, 29, 640–642. [Google Scholar] [CrossRef]
- Nagaishi, R.; Akehi, Y.; Ashida, K.; Higuchi-Tubouchi, K.; Yokoyama, H.; Nojiri, T.; Aoki, M.; Anzai, K.; Nabeshima, K.; Tanaka, M.; et al. Acute inflammatory syndrome and intrahepatic cholestasis caused by an interleukin-6-producing pheochromocytoma with pregnancy. Fukuoka Igaku Zasshi 2010, 101, 10–18. [Google Scholar]
- Ciacciarelli, M.; Bellini, D.; Laghi, A.; Polidoro, A.; Pacelli, A.; Bottaccioli, A.G.; Palmaccio, G.; Stefanelli, F.; Clemenzi, P.; Carini, L.; et al. IL-6-Producing, Noncatecholamines Secreting Pheochromocytoma Presenting as Fever of Unknown Origin. Case Rep. Med. 2016, 2016, 3489046. [Google Scholar] [CrossRef]
- Yarman, S.; Soyluk, O.; Altunoglu, E.; Tanakol, R. Interleukin-6-producing pheochromocytoma presenting with fever of unknown origin. Clinics (Sao Paulo) 2011, 66, 1843–1845. [Google Scholar] [CrossRef] [Green Version]
- Omura, M.; Sato, T.; Cho, R.; Iizuka, T.; Fujiwara, T.; Okamoto, K.; Tashiro, Y.; Chiba, S.; Nishikawa, T. A patient with malignant paraganglioma that simultaneously produces adrenocorticotropic hormone and interleukin-6. Cancer 1994, 74, 1634–1639. [Google Scholar] [CrossRef]
- Garbini, A.; Mainardi, M.; Grimi, M.; Repaci, G.; Nanni, G.; Bragherio, G. Pheochromocytoma and hypercalcemia due to ectopic production of parathyroid hormone. N. Y. State J. Med. 1986, 86, 25–27. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, M.; Xiao, Y.; Li, H.; Zhang, Y.; Ji, Z. Interleukin-6-producing pheochromocytoma as a new reason for fever of unknown origin: A retrospective study. Endocr. Pract. 2018, 24, 507–511. [Google Scholar] [CrossRef]
- Shanberg, A.M.; Baghdassarian, R.; Tansey, L.A.; Bacon, D.; Greenberg, P.; Perley, M. Pheochromocytoma with hypercalcemia: Case report and review of literature. J. Urol. 1985, 133, 258–259. [Google Scholar] [CrossRef]
- Mune, T.; Katakami, H.; Kato, Y.; Yasuda, K.; Matsukura, S.; Miura, K. Production and secretion of parathyroid hormone-related protein in pheochromocytoma: Participation of an alpha-adrenergic mechanism. J. Clin. Endocrinol. Metab. 1993, 76, 757–762. [Google Scholar]
- Bridgewater, J.A.; Ratcliffe, W.A.; Bundred, N.J.; Owens, C.W. Malignant phaeochromocytoma and hypercalcaemia. Postgrad. Med. J. 1993, 69, 77–79. [Google Scholar] [CrossRef]
- Kimura, S.; Nishimura, Y.; Yamaguchi, K.; Nagasaki, K.; Shimada, K.; Uchida, H. A case of pheochromocytoma producing parathyroid hormone-related protein and presenting with hypercalcemia. J. Clin. Endocrinol. Metab. 1990, 70, 1559–15563. [Google Scholar] [CrossRef]
- Fairhurst, B.J.; Shettar, S.P. Hypercalcaemia and phaeochromocytoma. Postgrad. Med. J. 1981, 57, 459–460. [Google Scholar] [CrossRef]
- Kanamori, A.; Suzuki, S.; Suzuki, Y.; Abe, Y.; Takada, I.; Fujita, Y.; Yajima, Y.; Okabe, H.; Kameya, T.; Yamaguchi, K. A case of pheochromocytoma associated with hypercalcitoninemia and ectopic production of many peptide hormones. Nippon Naika GakkaiZasshi 1986, 75, 1610–1615. [Google Scholar] [CrossRef]
- Heath, H., 3rd; Edis, A.J. Pheochromocytoma associated with hypercalcemia and ectopic secretion of calcitonin. Ann. Intern. Med. 1979, 91, 208–210. [Google Scholar] [CrossRef]
- Kalager, T.; Glück, E.; Heimann, P.; Myking, O. Phaeochromocytoma with ectopic calcitonin production and parathyroid cyst. Br. Med. J. 1977, 2, 21. [Google Scholar] [CrossRef]
- Vieira Neto, L.; Taboada, G.F.; Corrêa, L.L.; Polo, J.; Nascimento, A.F.; Chimelli, L.; Rumilla, K.; Gadelha, M.R. Acromegaly secondary to growth hormone-releasing hormone secreted by an incidentally discovered pheochromocytoma. Endocr. Pathol. 2007, 18, 46–52. [Google Scholar] [CrossRef]
- Saito, H.; Sano, T.; Yamasaki, R.; Mitsuhashi, S.; Hosoi, E.; Saito, S. Demonstration of biological activity of a growth hormone-releasing hormone-like substance produced by a pheochromocytoma. Acta Endocrinol. (Cph.) 1993, 129, 246–250. [Google Scholar] [CrossRef]
- Sano, T.; Saito, H.; Yamazaki, R.; Kameyama, K.; Ikeda, M.; Hosoi, E.; Hizawa, K.; Saito, S. Production of growth hormone-releasing factor in pheochromocytoma. N. Engl. J. Med. 1984, 311, 1520. [Google Scholar]
- Roth, K.A.; Wilson, D.M.; Eberwine, J.; Dorin, R.I.; Kovacs, K.; Bensch, K.G.; Hoffman, A.R. Acromegaly and pheochromocytoma: A multiple endocrine syndrome caused by a plurihormonal adrenal medullary tumor. J. Clin. Endocrinol. Metab. 1986, 63, 1421–1426. [Google Scholar] [CrossRef]
- Ghazi, A.A.; Amirbaigloo, A.; Dezfooli, A.A.; Saadat, N.; Ghazi, S.; Pourafkari, M.; Tirgari, F.; Dhall, D.; Bannykh, S.; Melmed, S.; et al. Ectopic acromegaly due to growth hormone releasing hormone. Endocrine 2013, 43, 293–302. [Google Scholar] [CrossRef]
- Uysal, M.; Temiz, S.; Gul, N.; Yarman, S.; Tanakol, R.; Kapran, Y. Hypoglycemia due to ectopic release of insulin from a paraganglioma. Horm. Res. 2007, 67, 292–295. [Google Scholar] [CrossRef]
- Hirai, H.; Midorikawa, S.; Suzuki, S.; Sasano, H.; Watanabe, T.; Satoh, H. Somatostatin-secreting Pheochromocytoma Mimicking Insulin-dependent Diabetes Mellitus. Intern. Med. 2016, 55, 2985–2991. [Google Scholar] [CrossRef] [Green Version]
- Kaslow, A.M.; Riquier-Brison, A.; Peti-Peterdi, J.; Shillingford, N.; HaDuong, J.; Venkatramani, R.; Gayer, C.P. An ectopic renin-secreting adrenal corticoadenoma in a child with malignant hypertension. Physiol. Rep. 2016, 4, 5. [Google Scholar] [CrossRef]
- Wang, F.; Tong, A.; Li, C.; Cui, Y.; Sun, J.; Song, A.; Li, Y. AZD8055 inhibits ACTH secretion in a case of bilateral ACTH-secreting pheochromocytoma. Oncol. Lett. 2018, 16, 4561–4566. [Google Scholar] [CrossRef]
- Chung, C.H.; Wang, C.H.; Tzen, C.Y.; Liu, C.P. Intrahepatic cholestasis as a paraneoplastic syndrome associated with pheochromocytoma. J. Endocrinol. Investig. 2005, 28, 175–179. [Google Scholar] [CrossRef]
- Bernini, M.; Bacca, A.; Casto, G.; Carli, V.; Cupisti, A.; Carrara, D.; Farnesi, I.; Barsotti, G.; Naccarato, A.G.; Bernini, G. A case of pheochromocytoma presenting as secondary hyperaldosteronism, hyperparathyroidism, diabetes and proteinuric renal disease. Nephrol. Dial. Transplant. 2011, 26, 1104–1107. [Google Scholar] [CrossRef] [Green Version]
- Teno, S.; Tanabe, A.; Nomura, K.; Demura, H. Acutely exacerbated hypertension and increased inflammatory signs due to radiation treatment for metastatic pheochromocytoma. Endocr. J. 1996, 43, 511–516. [Google Scholar] [CrossRef]
- Mutabagani, K.H.; Klopfenstein, K.J.; Hogan, M.J.; Caniano, D.A. Metastatic paraganglioma and paraneoplastic-induced anemia in an adolescent: Treatment with hepatic arterial chemoembolization. J. Pediatr. Hematol. Oncol. 1999, 21, 544–547. [Google Scholar] [CrossRef]
- Cho, Y.Y.; Kwak, M.K.; Lee, S.E.; Ahn, S.H.; Kim, H.; Suh, S.; Kim, B.J.; Song, K.H.; Koh, J.M.; Kim, J.H.; et al. A clinical prediction model to estimate the metastatic potential of pheochromocytoma/paraganglioma: ASES score. Surgery 2018, 164, 511–517. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Tischler, A.; de Krijger, R.R. Diagnostic tests and biomarkers for pheochromocytoma and extra-adrenal paraganglioma: From routine laboratory methods to disease stratification. Endocr. Pathol. 2012, 23, 4–14. [Google Scholar] [CrossRef]
- Maurice, J.B.; Troke, R.; Win, Z.; Ramachandran, R.; Al-Nahhas, A.; Naji, M.; Dhillo, W.; Meeran, K.; Goldstone, A.P.; Martin, N.M.; et al. A comparison of the performance of ⁶⁸Ga-DOTATATE PET/CT and ¹²³I-MIBG SPECT in the diagnosis and follow-up phaeochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1266–1270. [Google Scholar] [CrossRef]
- Tan, T.H.; Hussein, Z.; Saad, F.F.; Shuaib, I.L. Diagnostic Performance of (68)Ga-DOTATATE PET/CT, (18)F-FDG PET/CT and (131)I-MIBG Scintigraphy in Mapping Metastatic Pheochromocytoma and Paraganglioma. Nucl. Med. Mol. Imaging 2015, 49, 143–151. [Google Scholar] [CrossRef]
- Mundschenk, J.; Unger, N.; Schulz, S.; Höllt, V.; Schulz, S.; Steinke, R.; Lehnert, H. Somatostatin receptor subtypes in human pheochromocytoma: Subcellular expression pattern and functional relevance for octreotide scintigraphy. J. Clin. Endocrinol. Metab. 2003, 88, 5150–5157. [Google Scholar] [CrossRef]
- Niemeijer, N.D.; Alblas, G.; van Hulsteijn, L.T.; Dekkers, O.M.; Corssmit, E.P. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. (Oxf.) 2014, 81, 642–651. [Google Scholar] [CrossRef]
- Van Hulsteijn, L.T.; Niemeijer, N.D.; Dekkers, O.M.; Corssmit, E.P. (131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. (Oxf.) 2014, 80, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Feng, L.; Habra, M.A.; Rich, T.; Dickson, P.V.; Perrier, N.; Phan, A.; Waguespack, S.; Patel, S.; Jimenez, C. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: Insights from the largest single-institutional experience. Cancer 2012, 118, 2804–28012. [Google Scholar] [CrossRef] [PubMed]
- Jawed, I.; Velarde, M.; Därr, R.; Wolf, K.I.; Adams, K.; Venkatesan, A.M.; Balasubramaniam, S.; Poruchynsky, M.S.; Reynolds, J.C.; Pacak, K.; et al. Continued Tumor Reduction of Metastatic Pheochromocytoma/Paraganglioma Harboring Succinate Dehydrogenase Subunit B Mutations with Cyclical Chemotherapy. Cell Mol. Neurobiol. 2018, 38, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Forrer, F.; Riedweg, I.; Maecke, H.R.; Mueller-Brand, J. Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 334–340. [Google Scholar]
- Van Essen, M.; Krenning, E.P.; Kooij, P.P.; Bakker, W.H.; Feelders, R.A.; de Herder, W.W.; Wolbers, J.G.; Kwekkeboom, D.J. Effects of therapy with [177Lu-DOTA, Tyr3]octreotate in patients with paraganglioma, meningioma, small cell lung carcinoma, and melanoma. J. Nucl. Med. 2006, 47, 1599–1606. [Google Scholar] [PubMed]
- Nastos, K.; Cheung, V.T.F.; Toumpanakis, C.; Navalkissoor, S.; Quigley, A.M.; Caplin, M.; Khoo, B. Peptide Receptor Radionuclide Treatment and (131)I-MIBG in the management of patients with metastatic/progressive phaeochromocytomas and paragangliomas. J. Surg. Oncol. 2017, 115, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Grozinsky-Glasberg, S.; Hofman, M.S.; Callahan, J.; Meirovitz, A.; Maimon, O.; Pattison, D.A.; Gross, D.J.; Hicks, R.J. Efficacy of Peptide Receptor Radionuclide Therapy for Functional Metastatic Paraganglioma and Pheochromocytoma. J. Clin. Endocrinol. Metab. 2017, 102, 3278–3287. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Characteristics | |
|---|---|
| Number (PC) | 14 (7) |
| Female sex, n (%) | 8 (57%) |
| Median age (IQR), years | 45 (30) |
| Size primary tumor (cm) | 4.25 (4) |
| Sychronous/ Metachronous metastases | 7/7 |
| Functionality n, (%) | 10 (71%) |
| -normetanephrines | 3 |
| -metanephrine | 0 |
| -normetanephrines and metanephrines | 4 |
| -dopamine | 1 |
| -normetanephrine and dopamine | 1 |
| -normetanephrines. metanephrnes, dopamine | 1 |
| Functional imaging | |
| -Octreoscan (positive, %) | 4/7 (57%) |
| -68Gallium labelled octreotide (positive, %) | 3/3 (100%) |
| -18F-FGD-PET (positive, %) | 9/11 (82%) |
| -131I-MIBG (positive, %) | 8/10 (80%) |
| Follow-up, median (IQR, range), years | 6 (10, 1–14) |
| Treatment (any line) | |
| -Surgery | 12 (86%) |
| -PRTTs (131I-MIBG or 17Lu-Dotate) | 5 (35%) |
| -Chemotherapy | 7 (50%) |
| -Radiotherapy | 2 (14%) |
| Genetic status (n) | 7 |
| -SDHB+ | 2/7 |
| -SDHD+ | 1/7 |
| Mortality | 3 (21%) |
| Characteristics | N (%) |
|---|---|
| Primary tumor location, n (%) | |
| -PC | 7 (50%) |
| -PGL | 6 (46%) |
| bladder PGL | 1 |
| para-aortic PGL | 2 |
| paravertebral PGL | 1 |
| abdominal PGL | 2 |
| -PCs + PGLs | 1 |
| Primary tumor size, mm (median, IQR) | 4.25, 4 |
| Location of metastases | |
| -lymph nodes (abdominal/cervical) | 9 (69%) |
| -liver | 6 (46%) |
| -lung | 2 (15%) |
| -bones | 3 (23%) |
| Metastases per patient | >2 (1–4) |
| Histopathological data | |
| -Ki-67 (mean ± SD) | 11 ± 3.8% |
| -PASS | 7.75 |
| -SSTR2,5(positive/total (n)) | (3/5) |
| Hormone | No of Cases | PCs | PGLs | Malignant (n) | References |
|---|---|---|---|---|---|
| Total | 150 | 137 | 13 | 5 | |
| -ACTH | 49 | 43 | 6 | 2 (1PC, 1PGLs) | [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51] |
| -CRH | 8 | 6 | 2 | 0 | [52,53,54,55,56,57,58,59] |
| -VIP | 6 | 6 | 0 | 0 | [33,60,61,62] |
| -Vasopressin | 1 | 1 | 0 | 0 | [57] |
| -Calcium | 1 | 1 | 0 | 0 | [63] |
| -IL-6 | 40 | 39 | 1 | 1 | [64,65,66,67,68,69,70,71,72,73,74,75,76,77] |
| -PTH/PTHrp | 17 | 17 | 0 | 1 | [78,79,80,81,82] |
| -Calcitonin | 5 | 5 | 0 | 0 | [33,83,84,85] |
| -GH/GHRH | 7 | 6 | 1 | 0 | [86,87,88,89,90] |
| -Insuline/IGF-1 | 1 | 0 | 1 | 0 | [91] |
| -Somatostatin | 1 | 1 | 0 | 0 | [92] |
| -Aldosterone | 1 | 1 | 0 | 0 | [33] |
| -Renin | 2 | 2 | 0 | 0 | [33,93] |
| -CRH and ACTH | 2 | 1 | 1 | 0 | [59,94] |
| -CRH or ACTH and vasopressin | 2 | 2 | 0 | 0 | [57,58] |
| -IL b | 1 | 1 | 0 | [95] | |
| -ACTH and IL-6 | 1 | 0 | 1 | 1 | [76] |
| -Calcitonin and VIP | 3 | 3 | 0 | 0 | [33,63,83] |
| -PTH and aldosterone | 1 | 1 | 0 | 0 | [96] |
| -Neuropeptide Y | 1 | 1 | 0 | 0 | [16] |
| References | Number | PCs/PGLs | Metastases | Ectopic Secretion | Treatment |
|---|---|---|---|---|---|
| Kakudo K, et al. 1984 [25] | Case report (n = 1) | PC | Liver, lungs, bones, lymph nodes | ACTH (blood and tissue) | Surgery |
| Teno et al. 1996 [97] | Case report (n = 1) | PC | Bones | Suspicion of IL-6 but not measured | External Radiation |
| Tutal E et al. 2017 [30] | Case report (n = 1) | Renal PGL | Lymph nodes | ACTH (blood and tissues) | Surgery |
| Omura M et al. 1994 [75] | Case report (n = 1) | Cervical PGL | Bones | ACTH, IL-6 (blood and tumor) | Surgery and chemotherapy |
| Mutabagani KH et al. 1999 [98] | Case report (n = 1) | Mediastinal PGL | Liver | Anemia (probably Il-1 and anti-TNF secretion but never measured) | Surgery and hepatic arterial chemoembolization |
| Bridgewater JA et al. 1993 [80] | Case report (n = 1) | PC | Left para-aortic lymph node | PTHrp (blood and tissue) | Surgical resection |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelousi, A.; Peppa, M.; Chrisoulidou, A.; Alexandraki, K.; Berthon, A.; Faucz, F.R.; Kassi, E.; Kaltsas, G. Malignant Pheochromocytomas/Paragangliomas and Ectopic Hormonal Secretion: A Case Series and Review of the Literature. Cancers 2019, 11, 724. https://doi.org/10.3390/cancers11050724
Angelousi A, Peppa M, Chrisoulidou A, Alexandraki K, Berthon A, Faucz FR, Kassi E, Kaltsas G. Malignant Pheochromocytomas/Paragangliomas and Ectopic Hormonal Secretion: A Case Series and Review of the Literature. Cancers. 2019; 11(5):724. https://doi.org/10.3390/cancers11050724
Chicago/Turabian StyleAngelousi, Anna, Melpomeni Peppa, Alexandra Chrisoulidou, Krystallenia Alexandraki, Annabel Berthon, Fabio Rueda Faucz, Eva Kassi, and Gregory Kaltsas. 2019. "Malignant Pheochromocytomas/Paragangliomas and Ectopic Hormonal Secretion: A Case Series and Review of the Literature" Cancers 11, no. 5: 724. https://doi.org/10.3390/cancers11050724
APA StyleAngelousi, A., Peppa, M., Chrisoulidou, A., Alexandraki, K., Berthon, A., Faucz, F. R., Kassi, E., & Kaltsas, G. (2019). Malignant Pheochromocytomas/Paragangliomas and Ectopic Hormonal Secretion: A Case Series and Review of the Literature. Cancers, 11(5), 724. https://doi.org/10.3390/cancers11050724