Nerve Growth Factor Induces Proliferation and Aggressiveness in Prostate Cancer Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
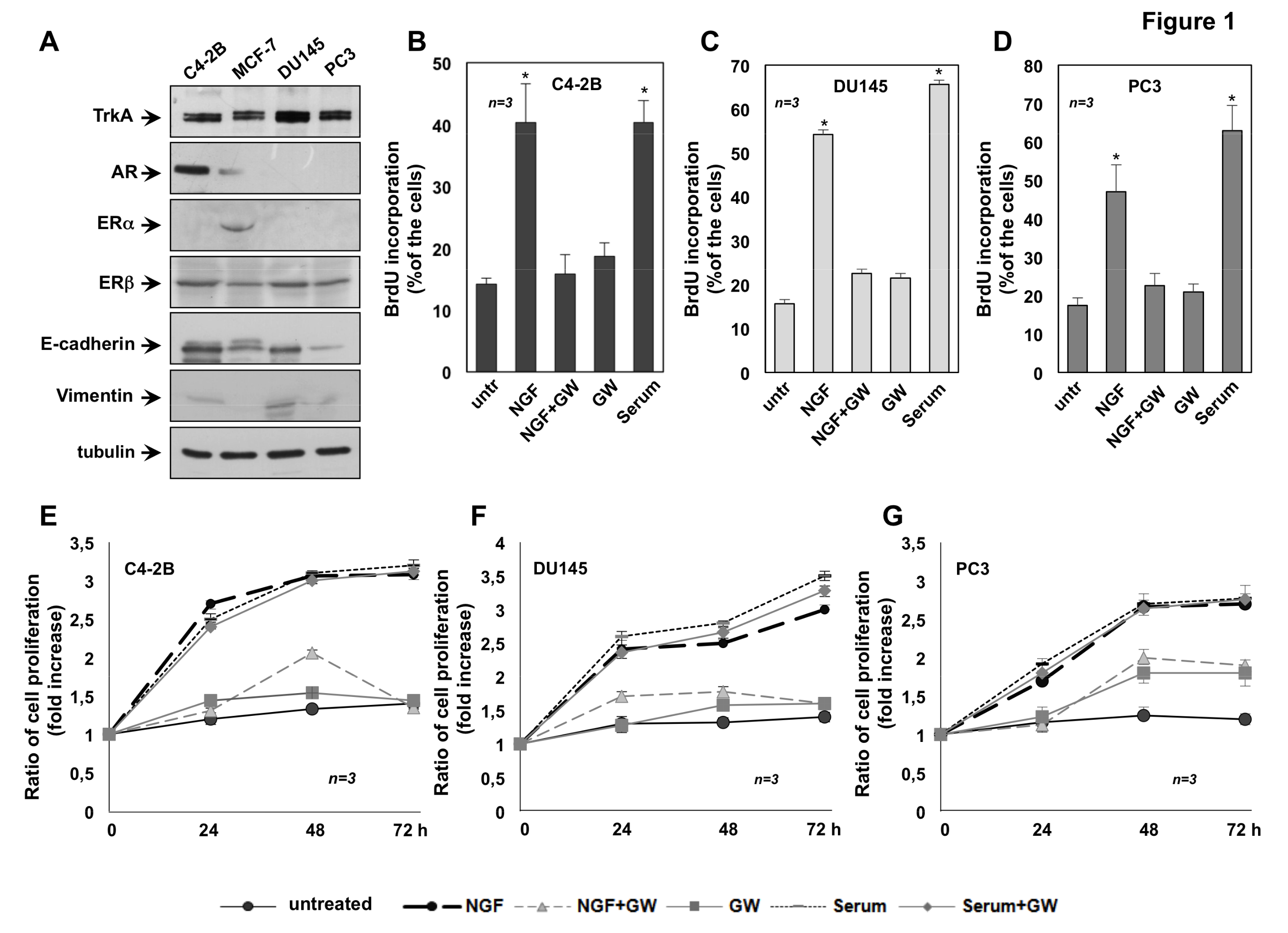
2.1. The Mitogenic Effect of NGF in CRPC Cells
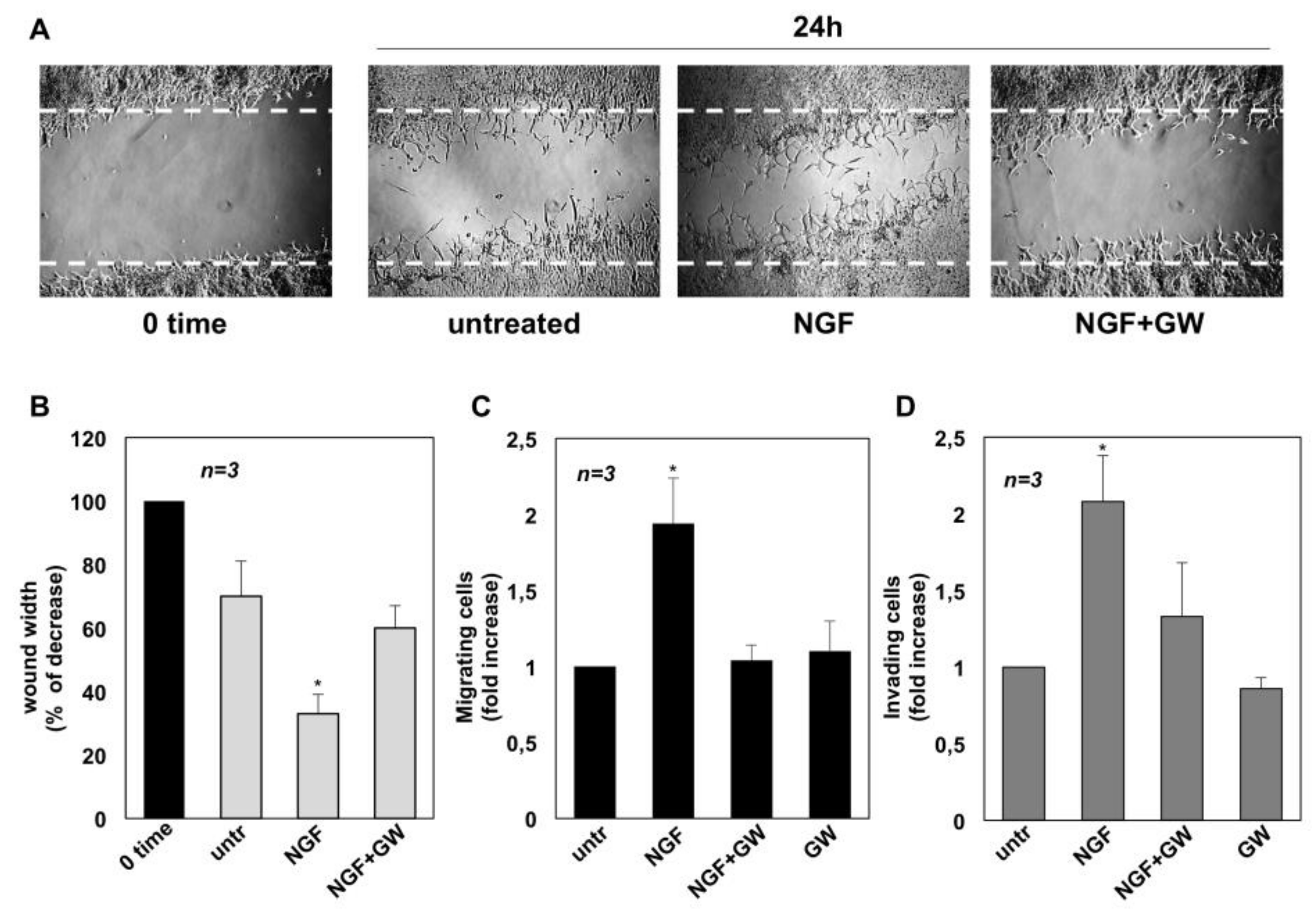
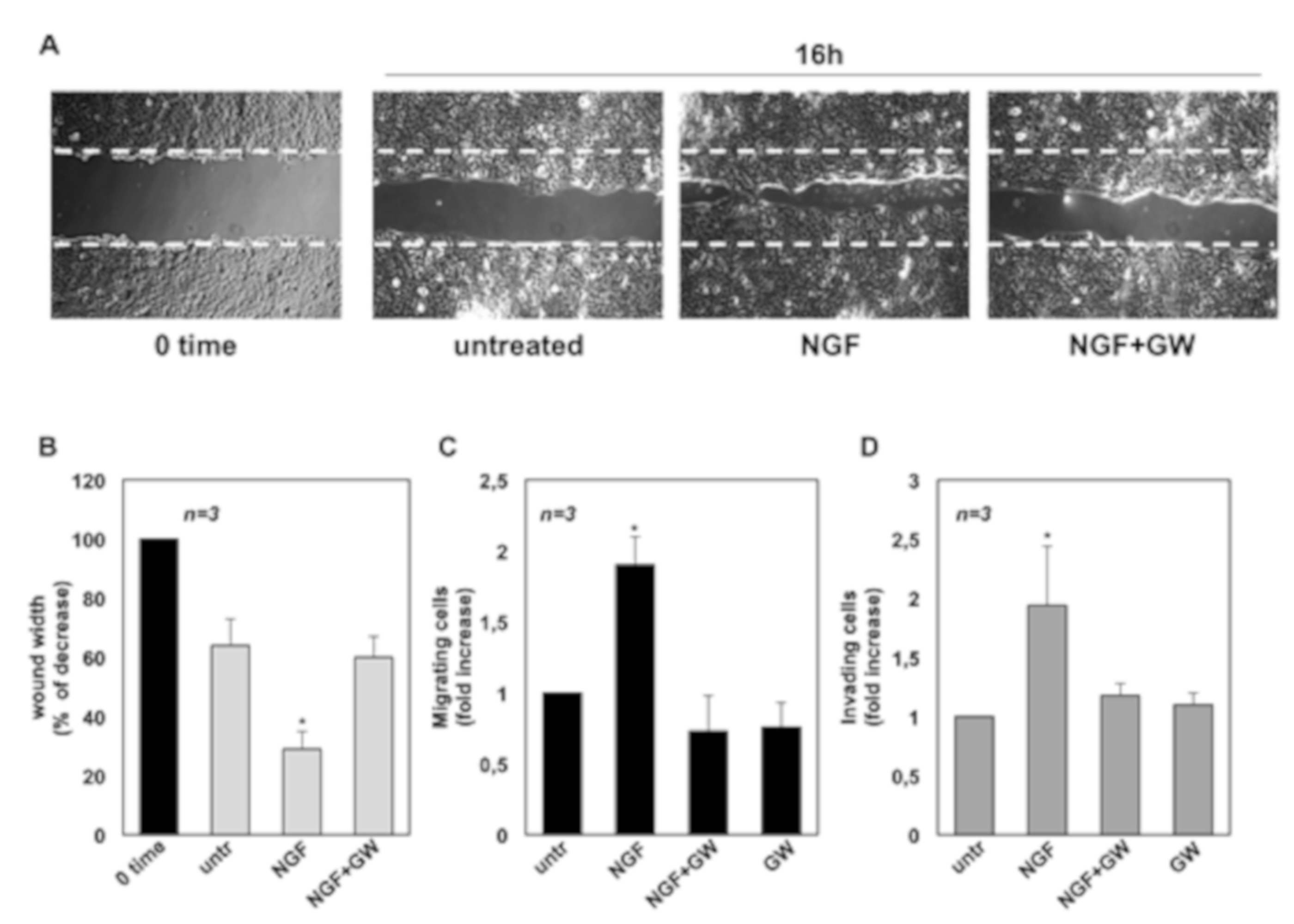
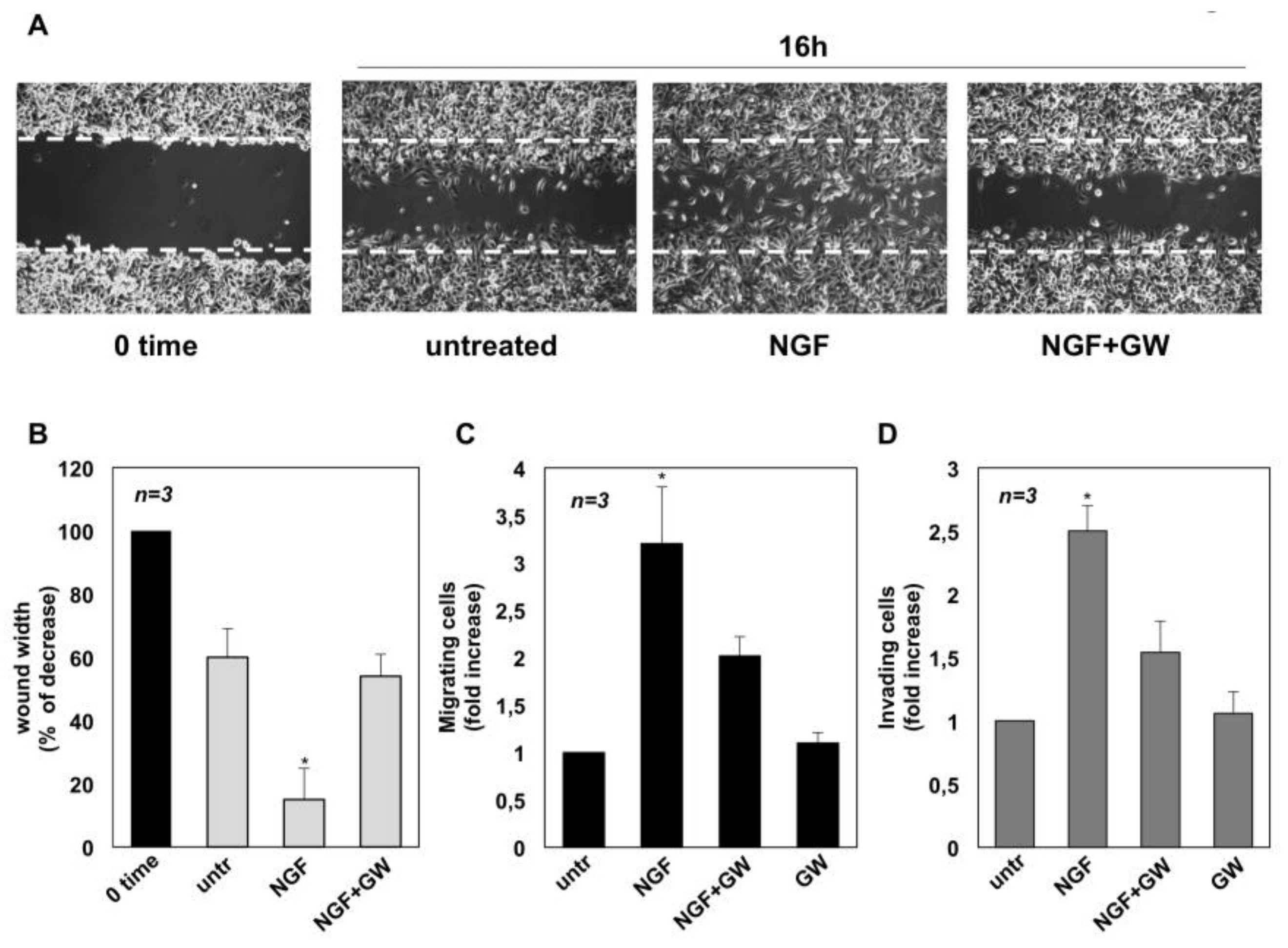
2.2. NGF Promotes Migration and Invasiveness of CRPC Cells Through TrkA Activation
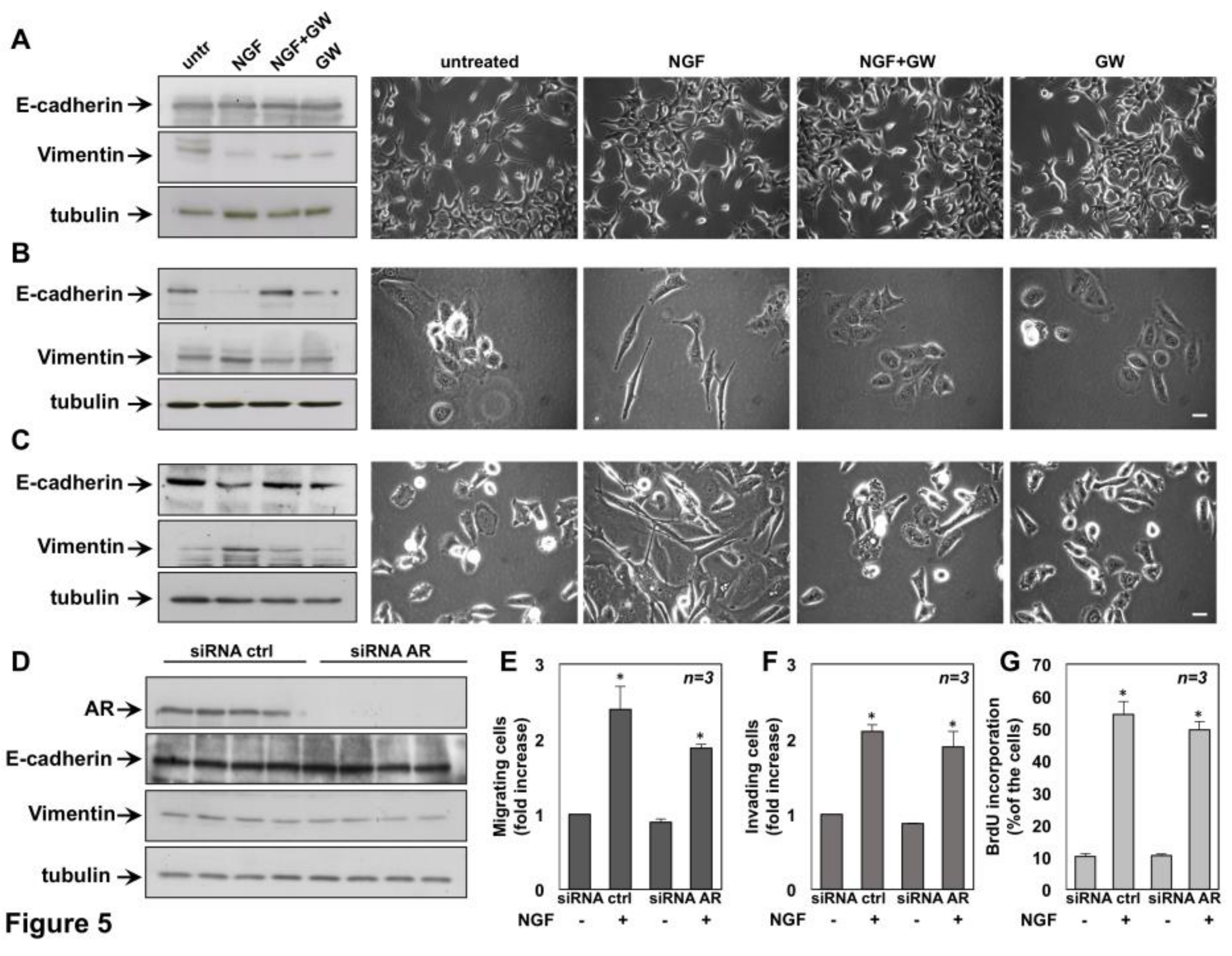
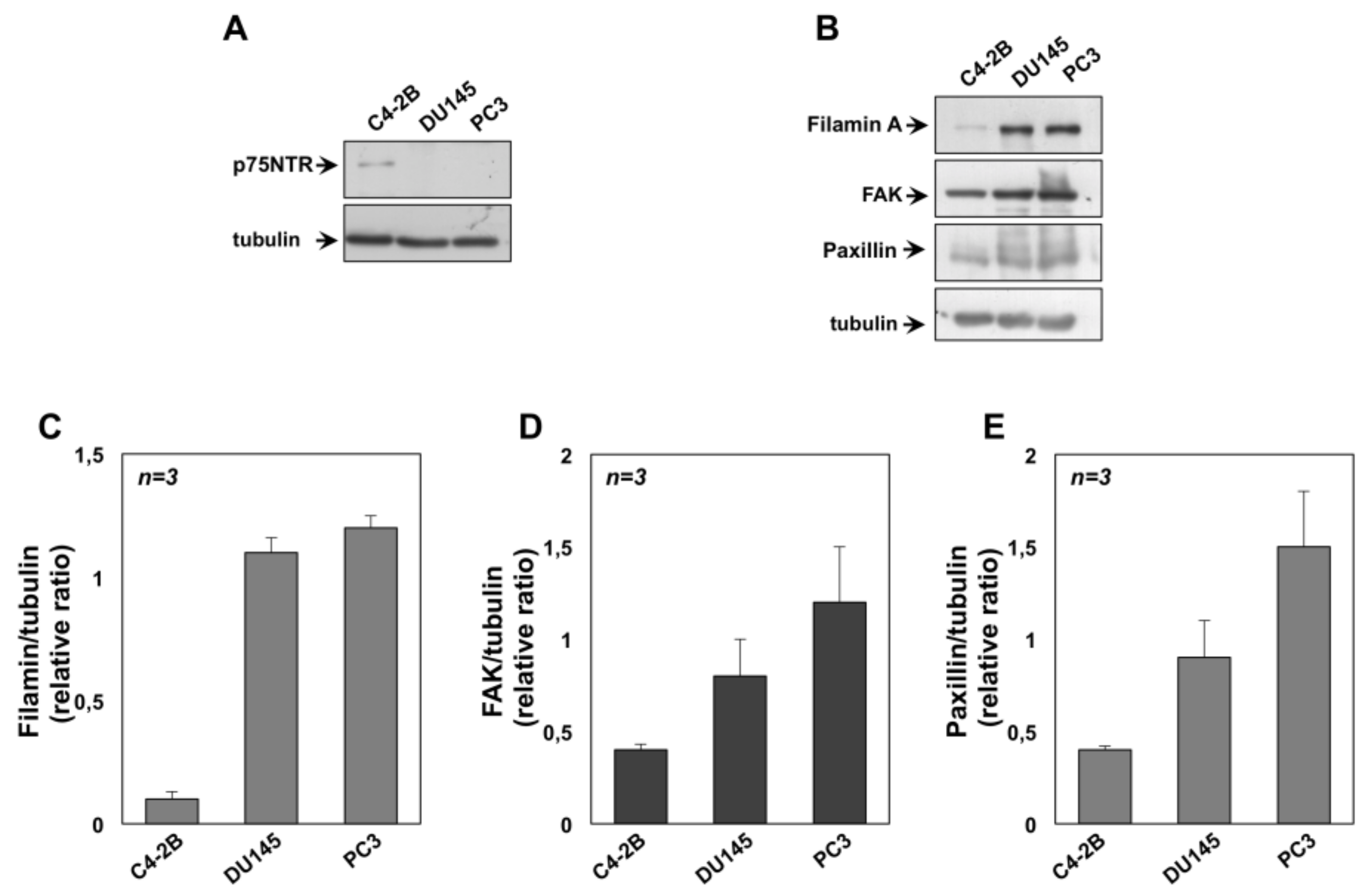
2.3. NGF Promotes EMT of DU145 and PC3 cells Through TrkA Activation
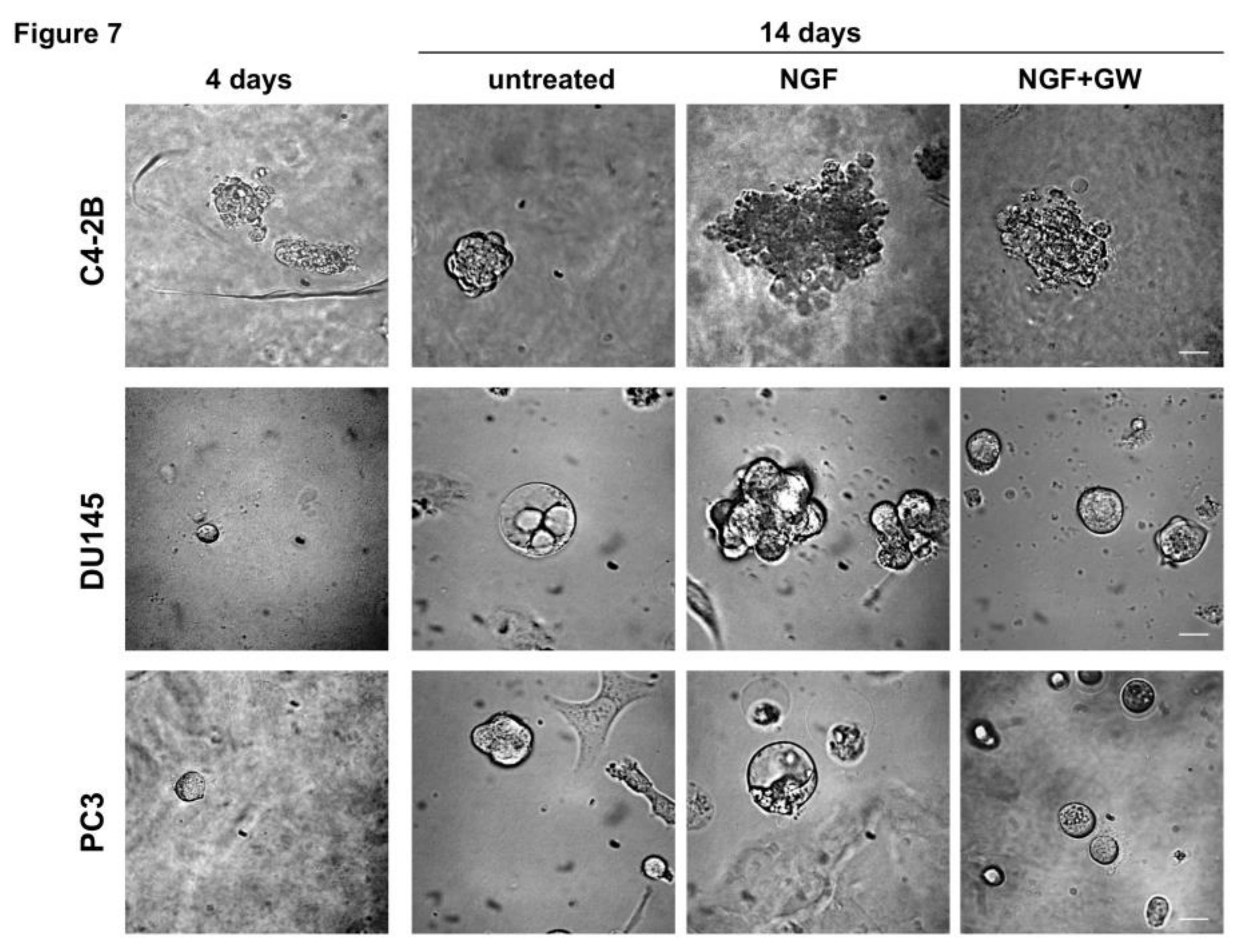
2.4. NGF Increases CRPC 3D-Organoid Growth Through TrkA Activation
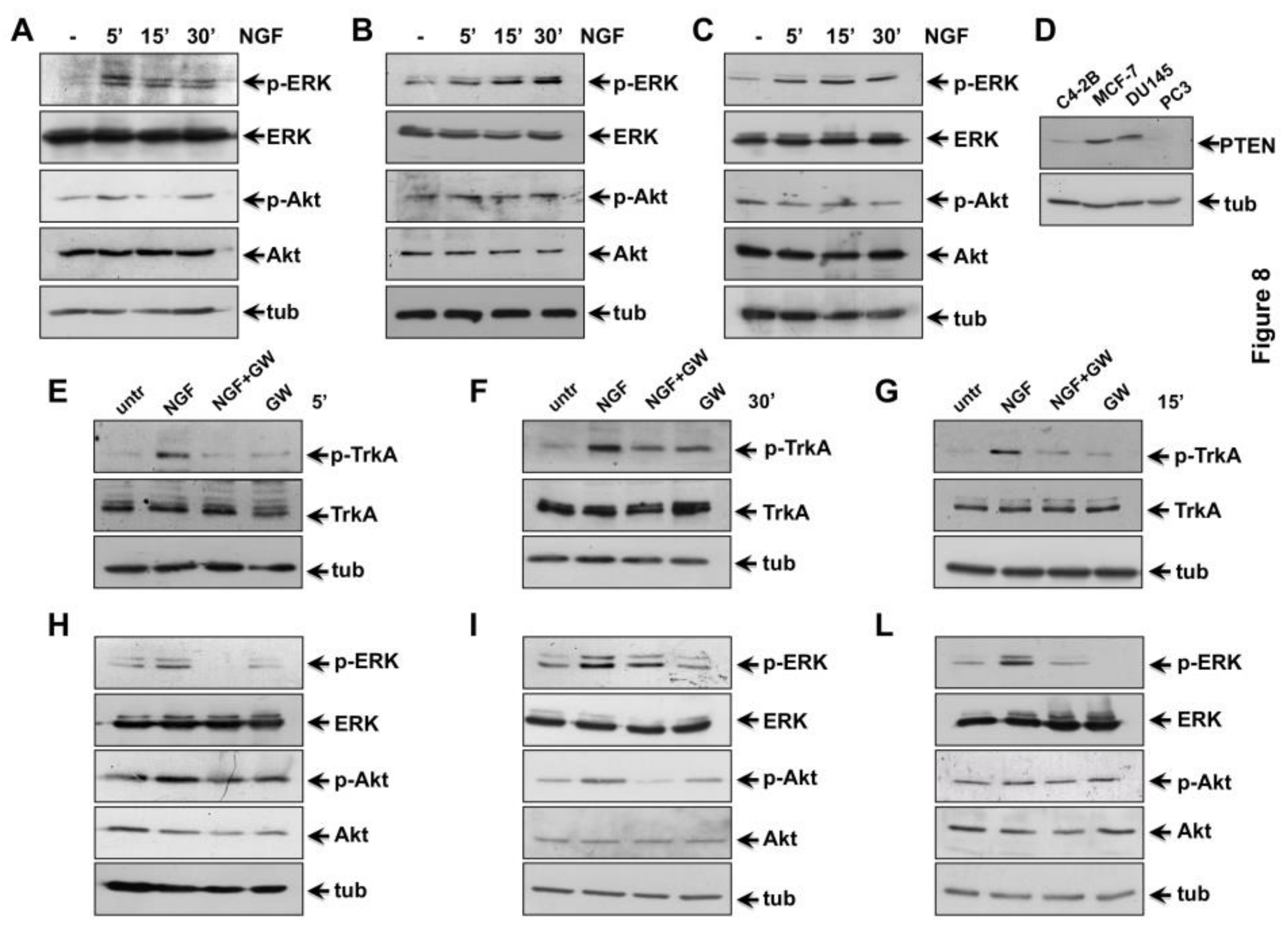
2.5. NGF Triggered TrkA-Tyr-490 Phosphorylation Controls MAPKs and Akt Activation in CRPC Cells
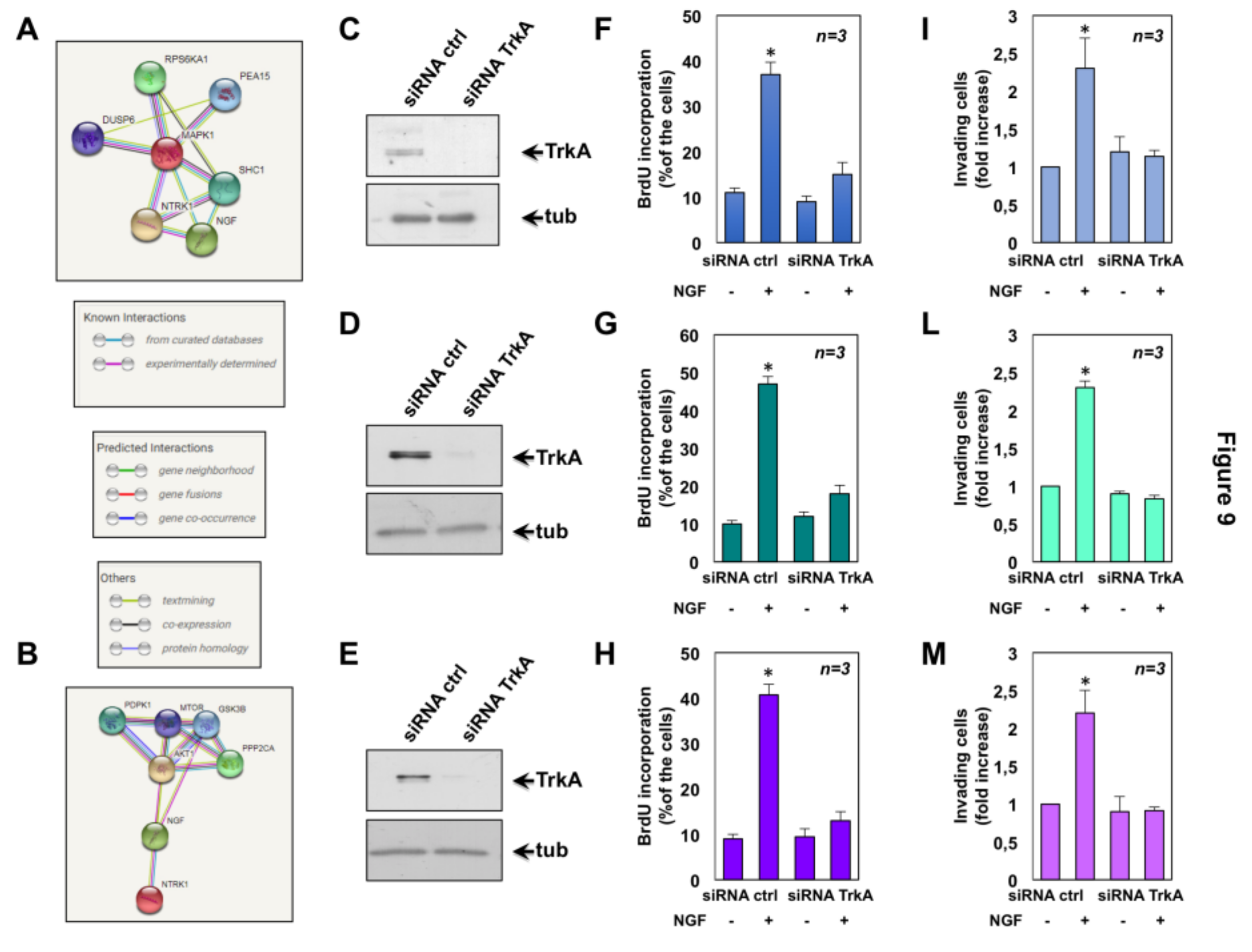
2.6. The Role of TrkA in NGF-Induced Proliferation and Invasiveness of CRPC Cells
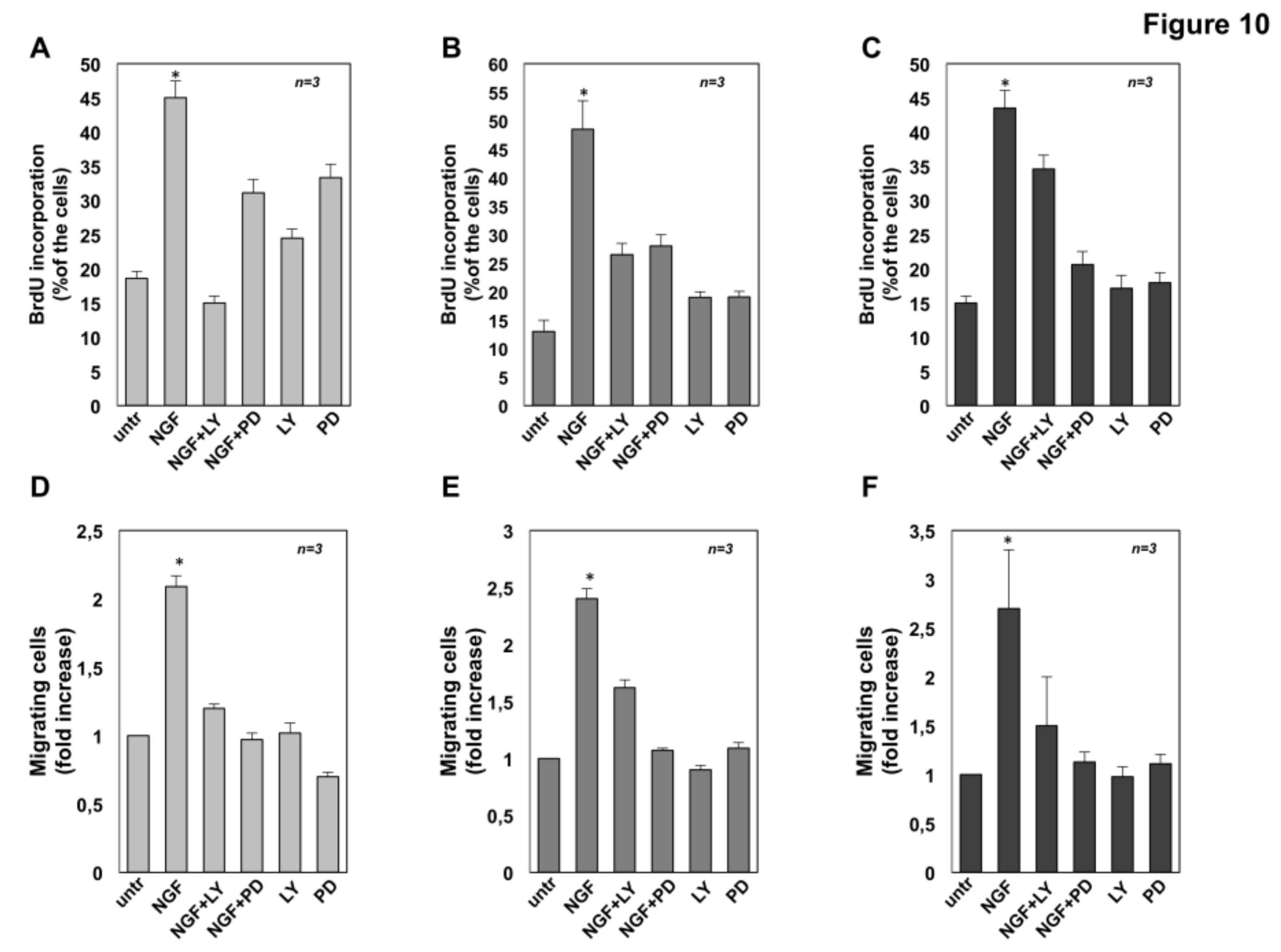
2.7. The Role of MAPK and Akt Activation in NGF-Induced Proliferation and Migration of CRPC Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Cultures
4.3. Transfection and siRNA experiments
4.4. DNA Synthesis, Immunofluorescence (IF) Microscopy and MTT Assay
4.5. Miniaturized 3D Cultures in ECM
4.6. Wound Scratch Analysis, Migration, Invasiveness and Phase-contrast Microscopy
4.7. EMT, Lysates and Western Blot Technique
4.8. Analysis of Network Construction
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Horwich, A.; Parker, C.; de Reijke, T.; Kataja, V. ESMO Guidelines Working Group Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24 (Suppl. 6), vi106–vi114. [Google Scholar]
- Leonel Almeida, P.; Jorge Pereira, B. Local Treatment of Metastatic Prostate Cancer: What is the Evidence So Far? Prostate Cancer 2018, 2018, 2654572. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.R.; Parsons, J.K.; Andriole, G.; Bahnson, R.R.; Barocas, D.A.; Castle, E.P.; Catalona, W.J.; Dahl, D.M.; Davis, J.W.; Epstein, J.I.; et al. NCCN Clinical Practice Guidelines Prostate Cancer Early Detection, Version 2.2015. J. Natl. Compr. Cancer Netw. 2015, 13, 1534–1561. [Google Scholar] [CrossRef]
- Ramamonjisoa, N.; Ackerstaff, E. Characterization of the Tumor Microenvironment and Tumor–Stroma Interaction by Non-invasive Preclinical Imaging. Front. Oncol. 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Bergeret, S.; Charbit, J.; Ansquer, C.; Bera, G.; Chanson, P.; Lussey-Lepoutre, C. Novel PET tracers: Added value for endocrine disorders. Endocrine 2019, 64, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Calabria, F.; Chiaravalloti, A.; Cicciò, C.; Gangemi, V.; Gullà, D.; Rocca, F.; Gallo, G.; Cascini, G.L.; Schillaci, O. PET/CT with 18F-choline: Physiological whole bio-distribution in male and female subjects and diagnostic pitfalls on 1000 prostate cancer patients: 18F-choline PET/CT bio-distribution and pitfalls. A southern Italian experience. Nucl. Med. Biol. 2017, 51, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Cuccurullo, V.; Di Stasio, G.D.; Evangelista, L.; Castoria, G.; Mansi, L. Biochemical and Pathophysiological Premises to Positron Emission Tomography With Choline Radiotracers. J. Cell. Physiol. 2017, 232, 270–275. [Google Scholar] [CrossRef]
- Rapozzi, V.; Ragno, D.; Guerrini, A.; Ferroni, C.; della Pietra, E.; Cesselli, D.; Castoria, G.; Di Donato, M.; Saracino, E.; Benfenati, V.; et al. Androgen Receptor Targeted Conjugate for Bimodal Photodynamic Therapy of Prostate Cancer in Vitro. Bioconjug. Chem. 2015, 26, 1662–1671. [Google Scholar] [CrossRef]
- Ahdoot, M.; Lebastchi, A.H.; Turkbey, B.; Wood, B.; Pinto, P.A. Contemporary treatments in prostate cancer focal therapy. Curr. Opin. Oncol. 2019, 31, 200–206. [Google Scholar] [CrossRef]
- Migliaccio, A.; Castoria, G.; de Falco, A.; Bilancio, A.; Giovannelli, P.; Di Donato, M.; Marino, I.; Yamaguchi, H.; Appella, E.; Auricchio, F. Polyproline and Tat transduction peptides in the study of the rapid actions of steroid receptors. Steroids 2012, 77, 974–978. [Google Scholar] [CrossRef]
- Tesei, A.; Leonetti, C.; Di Donato, M.; Gabucci, E.; Porru, M.; Varchi, G.; Guerrini, A.; Amadori, D.; Arienti, C.; Pignatta, S.; et al. Effect of small molecules modulating androgen receptor (SARMs) in human prostate cancer models. PLoS ONE 2013, 8, e62657. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, A.; Tesei, A.; Ferroni, C.; Paganelli, G.; Zamagni, A.; Carloni, S.; Di Donato, M.; Castoria, G.; Leonetti, C.; Porru, M.; et al. A new avenue toward androgen receptor pan-antagonists: C2 sterically hindered substitution of hydroxy-propanamides. J. Med. Chem. 2014, 57, 7263–7279. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.; Huang, B.; Gustaffson, J.A. Estrogen Receptor β as a Pharmaceutical Target. Trends Pharmacol. Sci. 2017, 38, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Di Santi, A.; Cernera, G.; Rossi, V.; Abbondanza, C.; Moncharmont, B.; Sinisi, A.A.; et al. Prostate cancer stem cells: The role of androgen and estrogen receptors. Oncotarget 2016, 7, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Castoria, G. Estrogens and Their Receptors in Prostate Cancer: Therapeutic Implications. Front Oncol. 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Levi-Montalcini, R. The nerve growth factor 35 years later. Science 1987, 237, 1154–1162. [Google Scholar] [CrossRef]
- Murphy, R.A.; Watson, A.Y.; Rhodes, J.A. Biological sources of nerve growth factor. Appl. Neurophysiol. 1984, 47, 33–42. [Google Scholar] [CrossRef]
- Cunha, G.R.; Ricke, W.; Thomson, A.; Marker, P.C.; Risbridger, G.; Hayward, S.W.; Wang, Y.Z.; Donjacour, A.A.; Kurita, T. Hormonal, cellular, and molecular regulation of normal and neoplastic prostatic development. J. Steroid Biochem. Mol. Biol. 2004, 92, 221–236. [Google Scholar] [CrossRef]
- Djakiew, D.; Pflug, B.; Onoda, M. Stromal-epithelial paracrine interactions in the neoplastic rat and human prostate. Adv. Exp. Med. Biol. 1993, 330, 185–202. [Google Scholar] [PubMed]
- Delsite, R.; Djakiew, D. Characterization of nerve growth factor precursor protein expression by human prostate stromal cells: A role in selective neurotrophin stimulation of prostate epithelial cell growth. Prostate 1999, 41, 39–48. [Google Scholar] [CrossRef]
- Graham, C.W.; Lynch, J.H.; Djakiew, D. Distribution of nerve growth factor-like protein and nerve growth factor receptor in human benign prostatic hyperplasia and prostatic adenocarcinoma. J. Urol. 1992, 147, 1444–1447. [Google Scholar] [CrossRef]
- Geldof, A.A.; De Kleijn, M.A.; Rao, B.R.; Newling, D.W. Nerve growth factor stimulates in vitro invasive capacity of DU145 human prostatic cancer cells. J. Cancer Res. Clin. Oncol. 1997, 123, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Walch, E.T.; Marchetti, D. Role of neurotrophins and neurotrophins receptors in the in vitro invasion and heparanase production of human prostate cancer cells. Clin. Exp. Metastasis 1999, 17, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Weeraratna, A.T.; Dalrymple, S.L.; Lamb, J.C.; Denmeade, S.R.; Miknyoczki, S.; Dionne, C.A.; Isaacs, J.T. Pan-trk inhibition decreases metastasis and enhances host survival in experimental models as a result of its selective induction of apoptosis of prostate cancer cells. Clin. Cancer Res. 2001, 7, 2237–2245. [Google Scholar] [PubMed]
- Festuccia, C.; Muzi, P.; Gravina, G.L.; Millimaggi, D.; Speca, S.; Dolo, V.; Ricevuto, E.; Vicentini, C.; Bologna, M. Tyrosine kinase inhibitor CEP-701 blocks the NTRK1/NGF receptor and limits the invasive capability of prostate cancer cells in vitro. Int. J. Oncol. 2007, 30, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Arrighi, N.; Bodei, S.; Zani, D.; Simeone, C.; Cunico, S.C.; Missale, C.; Spano, P.; Sigala, S. Nerve growth factor signaling in prostate health and disease. Growth Factors 2010, 28, 191–201. [Google Scholar] [CrossRef]
- Molloy, N.H.; Read, D.E.; Gorman, A.M. Nerve growth factor in cancer cell death and survival. Cancers (Basel) 2011, 3, 510–530. [Google Scholar] [CrossRef]
- Warrington, R.J.; Lewis, K.E. Natural antibodies against nerve growth factor inhibit in vitro prostate cancer cell metastasis. Cancer Immunol. Immunother. 2011, 60, 187–195. [Google Scholar] [CrossRef]
- Di Donato, M.; Bilancio, A.; D’Amato, L.; Claudiani, P.; Oliviero, M.A.; Barone, M.V.; Auricchio, A.; Appella, E.; Migliaccio, A.; Auricchio, F.; et al. Cross-talk between androgen receptor/filamin A and TrkA regulates neurite outgrowth in PC12 cells. Mol. Biol. Cell 2015, 26, 2858–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Donato, M.; Cernera, G.; Auricchio, F.; Migliaccio, A.; Castoria, G. Cross-talk between androgen receptor and nerve growth factor receptor in prostate cancer cells: Implications for a new therapeutic approach. Cell Death Discov. 2018, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Castoria, G.; Auricchio, F.; Migliaccio, A. Extranuclear partners of androgen receptor: At the crossroads of proliferation, migration, and neuritogenesis. FASEB J. 2017, 31, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- George, D.J.; Suzuki, H.; Bova, G.S.; Isaacs, J.T. Mutational analysis of the TrkA gene in prostate cancer. Prostate 1998, 36, 172–180. [Google Scholar] [CrossRef]
- Lange, A.M.; Lo, H.-W. Inhibiting TRK Proteins in Clinical Cancer Therapy. Cancers (Basel) 2018, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, G.N.; Anezinis, P.E.; Chang, S.M.; Zhau, H.E.; Kim, E.E.; Hopwood, V.L.; Pathak, S.; von Eschenbach, A.C.; Chung, L.W. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994, 54, 2577–2581. [Google Scholar] [PubMed]
- Thalmann, G.N.; Rhee, H.; Sikes, R.A.; Pathak, S.; Multani, A.; Zhau, H.E.; Marshall, F.F.; Chung, L.W.K. Human prostate fibroblasts induce growth and confer castration resistance and metastatic potential in LNCaP Cells. Eur. Urol. 2010, 58, 162–171. [Google Scholar] [CrossRef]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar]
- Stone, K.R.; Mickey, D.D.; Wunderli, H.; Mickey, G.H.; Paulson, D.F. Isolation of a human prostate carcinoma cell line (DU 145). Int. J. Cancer 1978, 21, 274–281. [Google Scholar] [CrossRef]
- Festuccia, C.; Gravina, G.L.; Muzi, P.; Pomante, R.; Ventura, L.; Ricevuto, E.; Vicentini, C.; Bologna, M. In vitro and in vivo effects of bicalutamide on the expression of TrkA and P75 neurotrophin receptors in prostate carcinoma. Prostate 2007, 67, 1255–1264. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Sokoll, L.J.; Dalrymple, S.; Rosen, D.M.; Gady, A.M.; Bruzek, D.; Ricklis, R.M.; Isaacs, J.T. Dissociation between androgen responsiveness for malignant growth vs. expression of prostate specific differentiation markers PSA, hK2, and PSMA in human prostate cancer models. Prostate 2003, 54, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Tilley, W.D.; Bentel, J.M.; Aspinall, J.O.; Hall, R.E.; Horsfall, D.J. Evidence for a novel mechanism of androgen resistance in the human prostate cancer cell line, PC-3. Steroids 1995, 60, 180–186. [Google Scholar] [CrossRef]
- Chlenski, A.; Nakashiro, K.; Ketels, K.V.; Korovaitseva, G.I.; Oyasu, R. Androgen receptor expression in androgen-independent prostate cancer cell lines. Prostate 2001, 47, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.J.; Kim, D.R. Apoptotic cell death in TrkA-overexpressing cells: Kinetic regulation of ERK phosphorylation and caspase-7 activation. Mol. Cells 2008, 26, 12–17. [Google Scholar] [PubMed]
- Wight, M. Insulin-like growth factor-(IGF-1) levels in bovine sera. Art Sci. 2000, 19, 2–3. [Google Scholar]
- Hugo, H.; Ackland, M.L.; Blick, T.; Lawrence, M.G.; Clements, J.A.; Williams, E.D.; Thompson, E.W. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J. Cell. Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Pflug, B.R.; Onoda, M.; Lynch, J.H.; Djakiew, D. Reduced expression of the low affinity nerve growth factor receptor in benign and malignant human prostate tissue and loss of expression in four human metastatic prostate tumor cell lines. Cancer Res. 1992, 52, 5403–5406. [Google Scholar]
- Pistore, C.; Giannoni, E.; Colangelo, T.; Rizzo, F.; Magnani, E.; Muccillo, L.; Giurato, G.; Mancini, M.; Rizzo, S.; Riccardi, M.; et al. DNA methylation variations are required for epithelial-to-mesenchymal transition induced by cancer-associated fibroblasts in prostate cancer cells. Oncogene 2017, 36, 5551–5566. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef]
- Härmä, V.; Virtanen, J.; Mäkelä, R.; Happonen, A.; Mpindi, J.-P.; Knuuttila, M.; Kohonen, P.; Lötjönen, J.; Kallioniemi, O.; Nees, M. A comprehensive panel of three-dimensional models for studies of prostate cancer growth, invasion and drug responses. PLoS ONE 2010, 5, e10431. [Google Scholar] [CrossRef] [PubMed]
- Descamps, S.; Toillon, R.A.; Adriaenssens, E.; Pawlowski, V.; Cool, S.M.; Nurcombe, V.; Le Bourhis, X.; Boilly, B.; Peyrat, J.P.; Hondermarck, H. Nerve growth factor stimulates proliferation and survival of human breast cancer cells through two distinct signaling pathways. J. Biol. Chem. 2001, 276, 17864–17870. [Google Scholar] [CrossRef] [PubMed]
- Gómez, N.; Cohen, P. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature 1991, 353, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, M.; Stephens, R.M.; Hallberg, B.; Downward, J.; Kaplan, D.R. The selective and inducible activation of endogenous PI 3-kinase in PC12 cells results in efficient NGF-mediated survival but defective neurite outgrowth. Oncogene 1999, 18, 4586–4597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.; Hope, J.M.; Guo, S.; Ong, Q.; François, A.; Kaplan, L.; Scherrer, G.; Cui, B. Optical Activation of TrkA Signaling. ACS Synth. Biol. 2018, 7, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Musatov, S.; Roberts, J.; Brooks, A.I.; Pena, J.; Betchen, S.; Pfaff, D.W.; Kaplitt, M.G. Inhibition of neuronal phenotype by PTEN in PC12 cells. Proc. Nat. Acad. Sci. USA 2004, 101, 3627–3631. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, D.R.; Martin-Zanca, D.; Parada, L.F. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature 1991, 350, 158–160. [Google Scholar] [CrossRef]
- Van Kanegan, M.J.; Strack, S. The protein phosphatase 2A regulatory subunits B’beta and B’delta mediate sustained TrkA neurotrophin receptor autophosphorylation and neuronal differentiation. Mol. Cell. Biol. 2009, 29, 662–674. [Google Scholar] [CrossRef]
- Obermeier, A.; Bradshaw, R.A.; Seedorf, K.; Choidas, A.; Schlessinger, J.; Ullrich, A. Neuronal differentiation signals are controlled by nerve growth factor receptor/Trk binding sites for SHC and PLC gamma. EMBO J. 1994, 13, 1585–1590. [Google Scholar] [CrossRef]
- Nakagawara, A. Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Lett. 2001, 169, 107–114. [Google Scholar] [CrossRef]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef] [PubMed]
- Spear, N.; Estévez, A.G.; Barbeito, L.; Beckman, J.S.; Johnson, G.V. Nerve growth factor protects PC12 cells against peroxynitrite-induced apoptosis via a mechanism dependent on phosphatidylinositol 3-kinase. J. Neurochem. 1997, 69, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Shimoke, K.; Kishi, S.; Utsumi, T.; Shimamura, Y.; Sasaya, H.; Oikawa, T.; Uesato, S.; Ikeuchi, T. NGF-induced phosphatidylinositol 3-kinase signaling pathway prevents thapsigargin-triggered ER stress-mediated apoptosis in PC12 cells. Neurosci. Lett. 2005, 389, 124–128. [Google Scholar] [CrossRef]
- Wang, C.; Cigliano, A.; Delogu, S.; Armbruster, J.; Dombrowski, F.; Evert, M.; Chen, X.; Calvisi, D.F. Functional crosstalk between AKT/mTOR and Ras/MAPK pathways in hepatocarcinogenesis. Cell Cycle 2013, 12, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Bostwick, D.G.; Burke, H.B.; Djakiew, D.; Euling, S.; Ho, S.M.; Landolph, J.; Morrison, H.; Sonawane, B.; Shifflett, T.; Waters, D.J.; et al. Human prostate cancer risk factors. Cancer 2004, 101, 2371–2490. [Google Scholar] [CrossRef] [PubMed]
- Djakiew, D. Dysregulated expression of growth factors and their receptors in the development of prostate cancer. Prostate 2000, 42, 150–160. [Google Scholar] [CrossRef]
- Krygier, S.; Djakiew, D. Neurotrophin receptor p75(NTR) suppresses growth and nerve growth factor-mediated metastasis of human prostate cancer cells. Int. J. Cancer 2002, 98, 1–7. [Google Scholar] [CrossRef]
- Krygier, S.; Djakiew, D. Molecular characterization of the loss of p75(NTR) expression in human prostate tumor cells. Mol Carcinog. 2001, 31, 46–55. [Google Scholar] [CrossRef]
- Weeraratna, A.T.; Arnold, J.T.; George, D.J.; DeMarzo, A.; Isaacs, J.T. Rational basis for Trk inhibition therapy for prostate cancer. Prostate 2000, 45, 140–148. [Google Scholar] [CrossRef]
- Dalal, R.; Djakiew, D. Molecular characterization of neurotrophin expression and the corresponding tropomyosin receptor kinases (trks) in epithelial and stromal cells of the human prostate. Mol. Cell. Endocrinol. 1997, 134, 15–22. [Google Scholar] [CrossRef]
- Sampson, N.; Neuwirt, H.; Puhr, M.; Klocker, H.; Eder, I.E. In vitro model systems to study androgen receptor signaling in prostate cancer. Endocr. Relat. cancer 2013, 20, R49–R64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, C.M.; Gao, A.C. Current strategies for targeting the activity of androgen receptor variants. Asian J. Urol. 2019, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, D.; Somarelli, J.A.; Hanna, G.; Palmer, G.M.; Garcia-Blanco, M.A. Cellular migration and invasion uncoupled: Increased migration is not an inexorable consequence of epithelial-to-mesenchymal transition. Mol. Cell. Biol. 2014, 34, 3486–3499. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Walsh, C.A. The many faces of filamin: A versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 2004, 6, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Deakin, N.O.; Turner, C.E. Paxillin comes of age. J. Cell Sci. 2008, 121, 2435–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spans, L.; Helsen, C.; Clinckemalie, L.; Van den Broeck, T.; Prekovic, S.; Joniau, S.; Lerut, E.; Claessens, F. Comparative Genomic and Transcriptomic Analyses of LNCaP and C4-2B Prostate Cancer Cell Lines. PLoS ONE 2014, 9, e90002. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.; Domenico, M.D.; Castoria, G.; Nanayakkara, M.; Lombardi, M.; de Falco, A.; Bilancio, A.; Varricchio, L.; Ciociola, A.; Auricchio, F. Steroid Receptor Regulation of Epidermal Growth Factor Signaling through Src in Breast and Prostate Cancer Cells: Steroid Antagonist Action. Cancer Res. 2005, 65, 10585–10593. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Guo, Z.; Sun, F.; Li, W.; Alfano, A.; Shimelis, H.; Chen, M.; Brodie, A.M.H.; Chen, H.; Xiao, Z.; et al. Novel membrane-associated androgen receptor splice variant potentiates proliferative and survival responses in prostate cancer cells. J. Biol. Chem. 2011, 286, 36152–36160. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Karri, D.; Shen, H.; Shao, J.; Dasgupta, S.; Huang, S.; Edwards, D.P.; Ittmann, M.M.; O’Malley, B.W.; Yi, P. TRAF4-mediated ubiquitination of NGF receptor TrkA regulates prostate cancer metastasis. J. Clin. Investig. 2018, 128, 3129–3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, C.; Carducci, M.A.; Eisenberger, M.A.; Isaacs, J.T.; Partin, A.W.; Pili, R.; Sinibaldi, V.J.; Walczak, J.S.; Denmeade, S.R. Preclinical and clinical studies with the multi-kinase inhibitor CEP-701 as treatment for prostate cancer demonstrate the inadequacy of PSA response as a primary endpoint. Cancer Biol. Ther. 2007, 6, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Dionne, C.A.; Camoratto, A.M.; Jani, J.P.; Emerson, E.; Neff, N.; Vaught, J.L.; Murakata, C.; Djakiew, D.; Lamb, J.; Bova, S.; et al. Cell cycle-independent death of prostate adenocarcinoma is induced by the trk tyrosine kinase inhibitor CEP-751 (KT6587). Clin. Cancer Res. 1998, 4, 1887–1898. [Google Scholar] [PubMed]
- Bilancio, A.; Migliaccio, A. Phosphoinositide 3-Kinase Assay in Breast Cancer Cell Extracts. In Steroid Receptors: Methods and Protocols; Castoria, G., Auricchio, F., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; pp. 145–153. ISBN 978-1-4939-1346-6. [Google Scholar]
- Castoria, G.; Giovannelli, P.; Di Donato, M.; Ciociola, A.; Hayashi, R.; Bernal, F.; Appella, E.; Auricchio, F.; Migliaccio, A. Role of non-genomic androgen signalling in suppressing proliferation of fibroblasts and fibrosarcoma cells. Cell Death Dis. 2014, 5, e1548. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Naviglio, S.; Spina, A.; Chiosi, E.; Castoria, G.; Romano, M.; Sorrentino, A.; Illiano, F.; Illiano, G. Differentiation of H9c2 cardiomyoblasts: The role of adenylate cyclase system. J. Cell Physiol. 2004, 198, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Bilancio, A.; Bontempo, P.; Di Donato, M.; Conte, M.; Giovannelli, P.; Altucci, L.; Migliaccio, A.; Castoria, G. Bisphenol A induces cell cycle arrest in primary and prostate cancer cells through EGFR/ERK/p53 signaling pathway activation. Oncotarget 2017, 8, 115620–115631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, C.W.; Shibata, M.; Lei, M.; Toivanen, R.; Barlow, L.J.; Bergren, S.K.; Badani, K.K.; McKiernan, J.M.; Benson, M.C.; Hibshoosh, H.; et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat. Cell Biol. 2014, 16, 951. [Google Scholar] [CrossRef]
- Beshiri, M.L.; Tice, C.M.; Tran, C.; Nguyen, H.M.; Sowalsky, A.G.; Agarwal, S.; Jansson, K.H.; Yang, Q.; McGowen, K.M.; Yin, J.; et al. A PDX/Organoid Biobank of Advanced Prostate Cancers Captures Genomic and Phenotypic Heterogeneity for Disease Modeling and Therapeutic Screening. Clin. Cancer Res. 2018, 24, 4332–4345. [Google Scholar] [CrossRef] [Green Version]
- Castoria, G.; D’Amato, L.; Ciociola, A.; Giovannelli, P.; Giraldi, T.; Sepe, L.; Paolella, G.; Barone, M.V.; Migliaccio, A.; Auricchio, F. Androgen-induced cell migration: Role of androgen receptor/filamin A association. PLoS ONE 2011, 6, e17218. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, G.; Migliaccio, A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells Through AR/Src/PI3-K Complex Assembly. Sci. Rep. 2019, 9, 4490. [Google Scholar] [CrossRef] [PubMed]
- Rossi, V.; Di Zazzo, E.; Galasso, G.; De Rosa, C.; Abbondanza, C.; Sinisi, A.A.; Altucci, L.; Migliaccio, A.; Castoria, G. Estrogens Modulate Somatostatin Receptors Expression and Synergize With the Somatostatin Analog Pasireotide in Prostate Cells. Front. Pharmacol. 2019, 10, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Donato, M.; Cernera, G.; Migliaccio, A.; Castoria, G. Nerve Growth Factor Induces Proliferation and Aggressiveness in Prostate Cancer Cells. Cancers 2019, 11, 784. https://doi.org/10.3390/cancers11060784
Di Donato M, Cernera G, Migliaccio A, Castoria G. Nerve Growth Factor Induces Proliferation and Aggressiveness in Prostate Cancer Cells. Cancers. 2019; 11(6):784. https://doi.org/10.3390/cancers11060784
Chicago/Turabian StyleDi Donato, Marzia, Gustavo Cernera, Antimo Migliaccio, and Gabriella Castoria. 2019. "Nerve Growth Factor Induces Proliferation and Aggressiveness in Prostate Cancer Cells" Cancers 11, no. 6: 784. https://doi.org/10.3390/cancers11060784
APA StyleDi Donato, M., Cernera, G., Migliaccio, A., & Castoria, G. (2019). Nerve Growth Factor Induces Proliferation and Aggressiveness in Prostate Cancer Cells. Cancers, 11(6), 784. https://doi.org/10.3390/cancers11060784