Inhibition of DNA Repair Mechanisms and Induction of Apoptosis in Triple Negative Breast Cancer Cells Expressing the Human Herpesvirus 6 U94


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
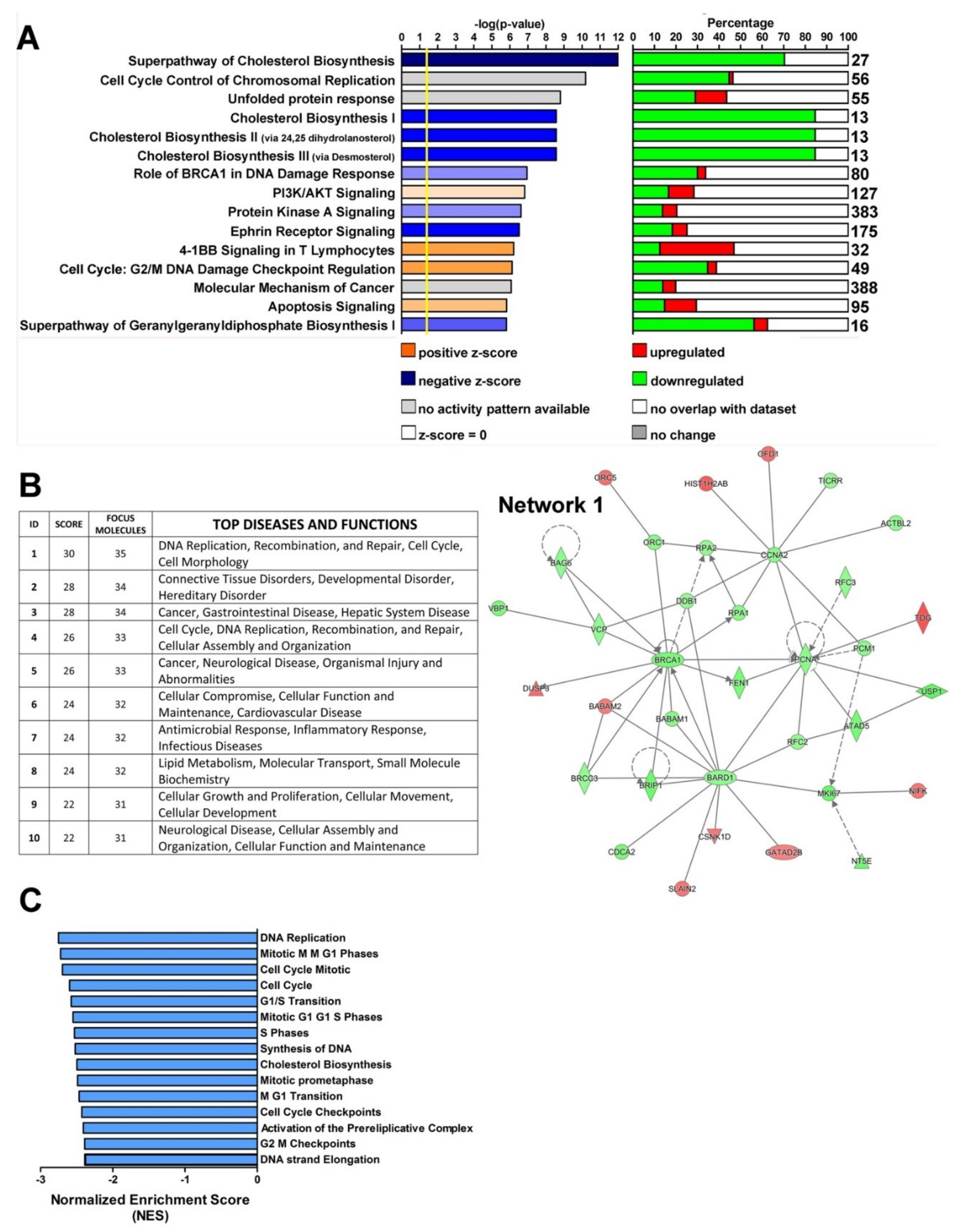
2.1. U94 Alters the Transcriptome Profile of MDA-MB 231 Cells
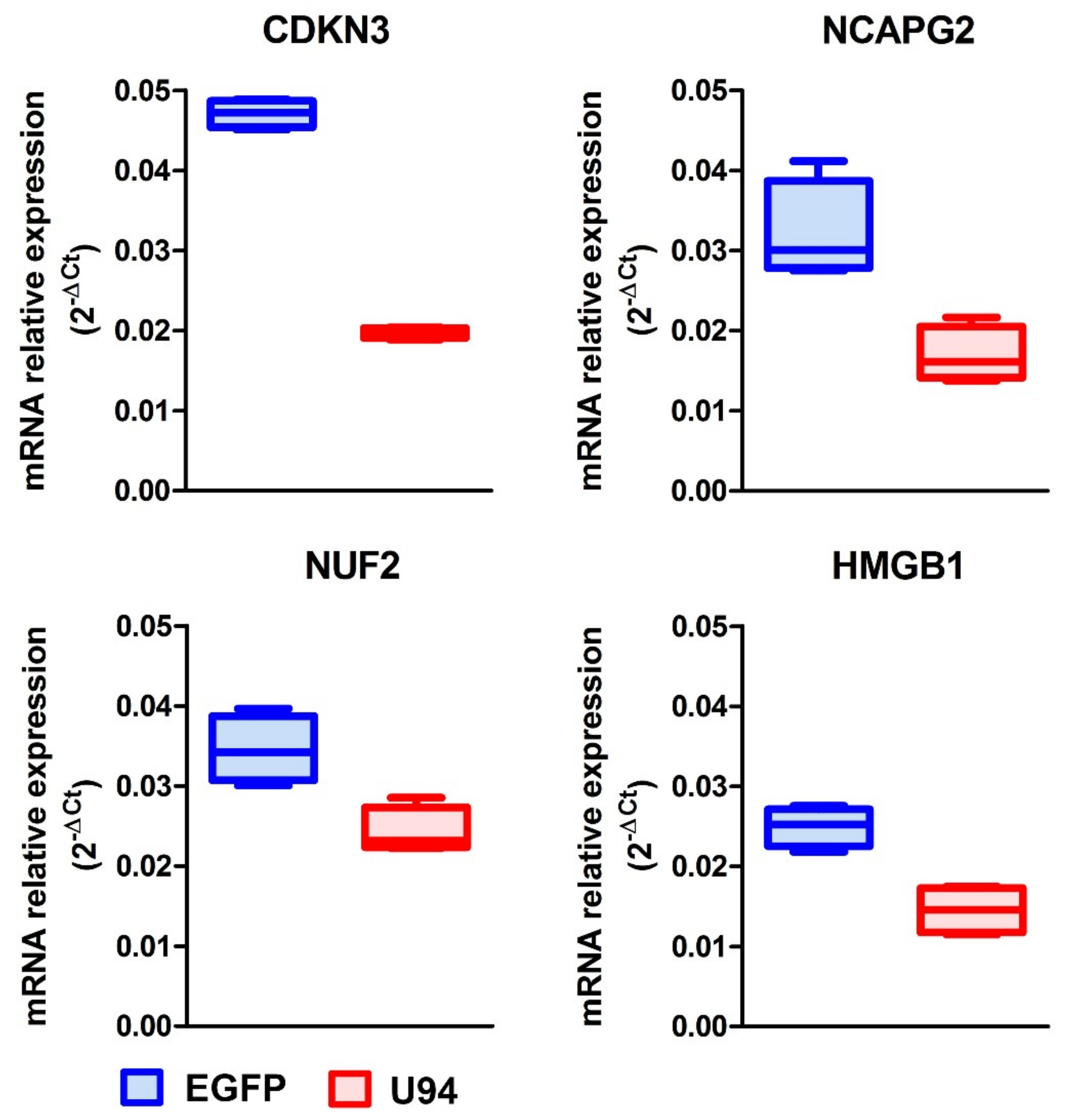
2.2. U94 Induces Downregulation of Specific Gene sets in MDA-MB 231 Breast Cancer Cells
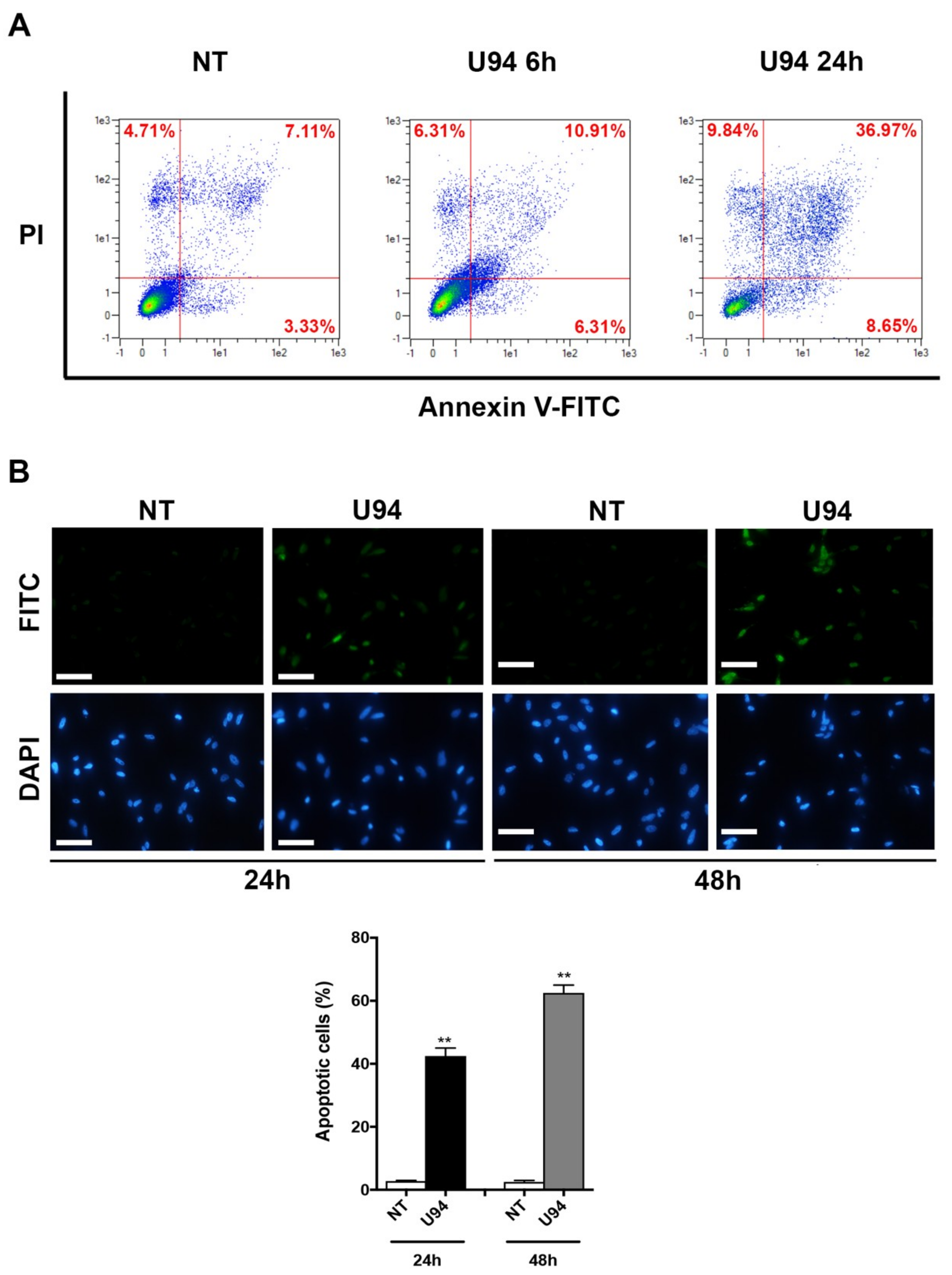
2.3. U94 Induces Breast Cancer Cell Apoptosis
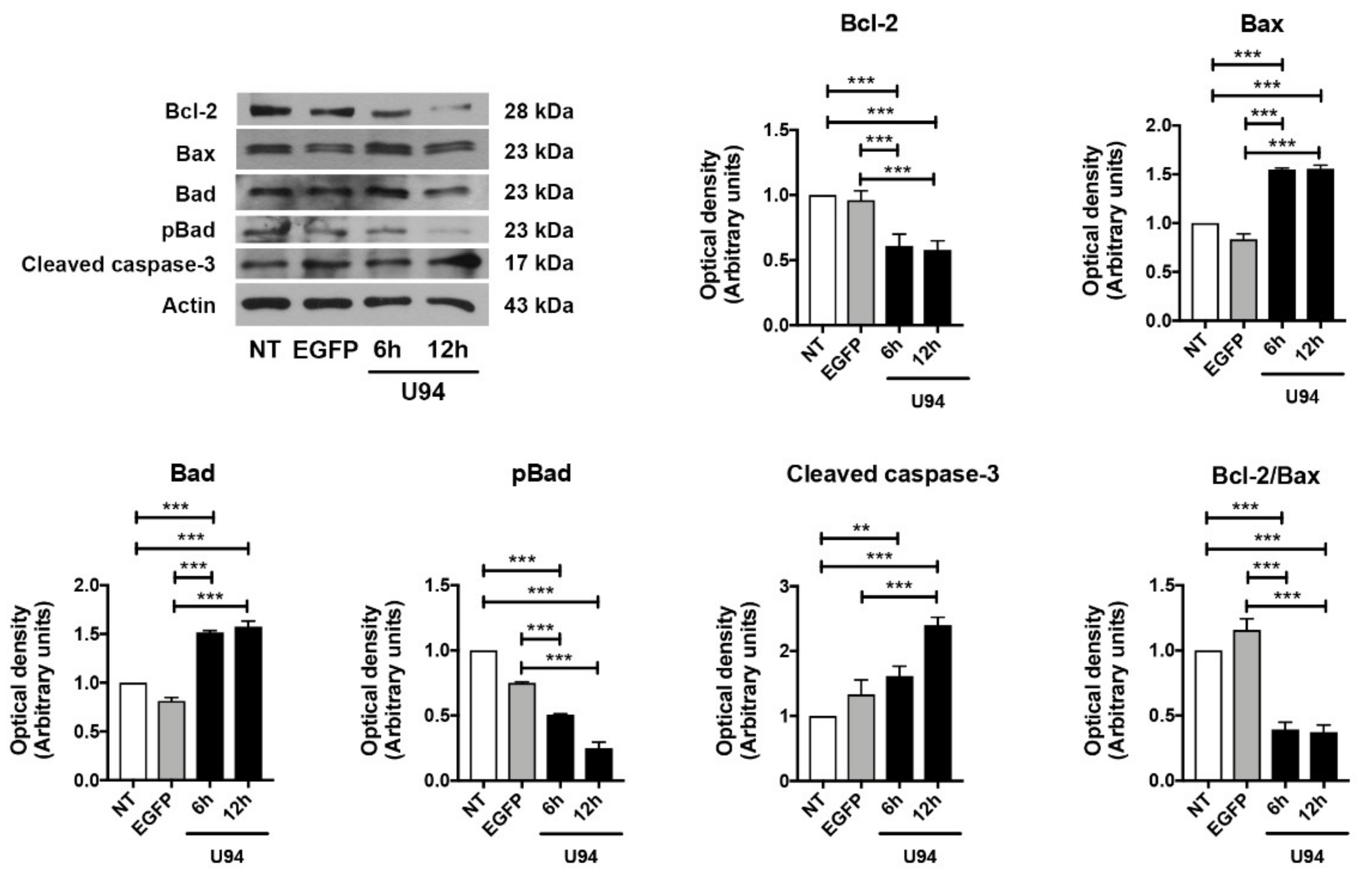
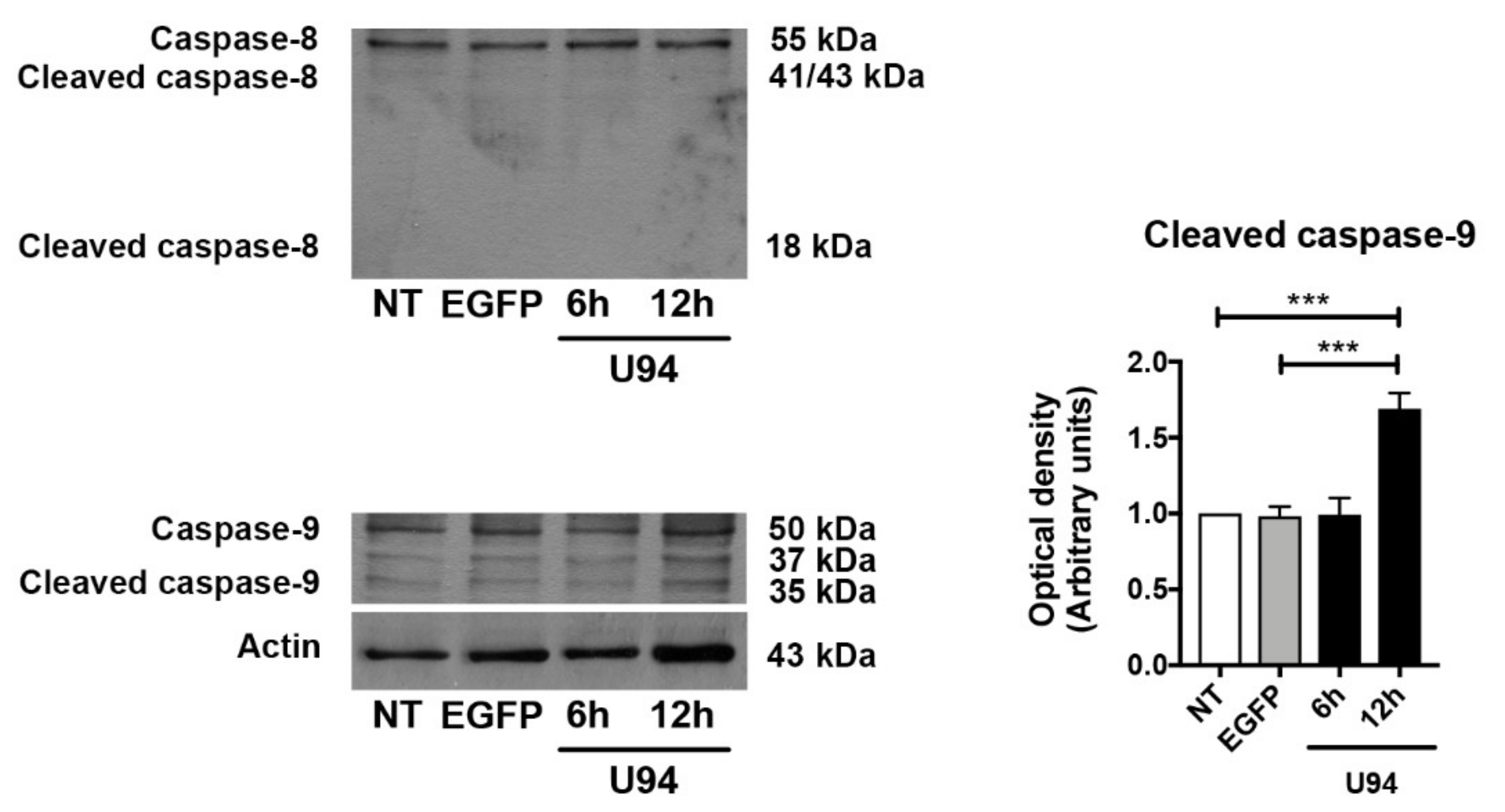
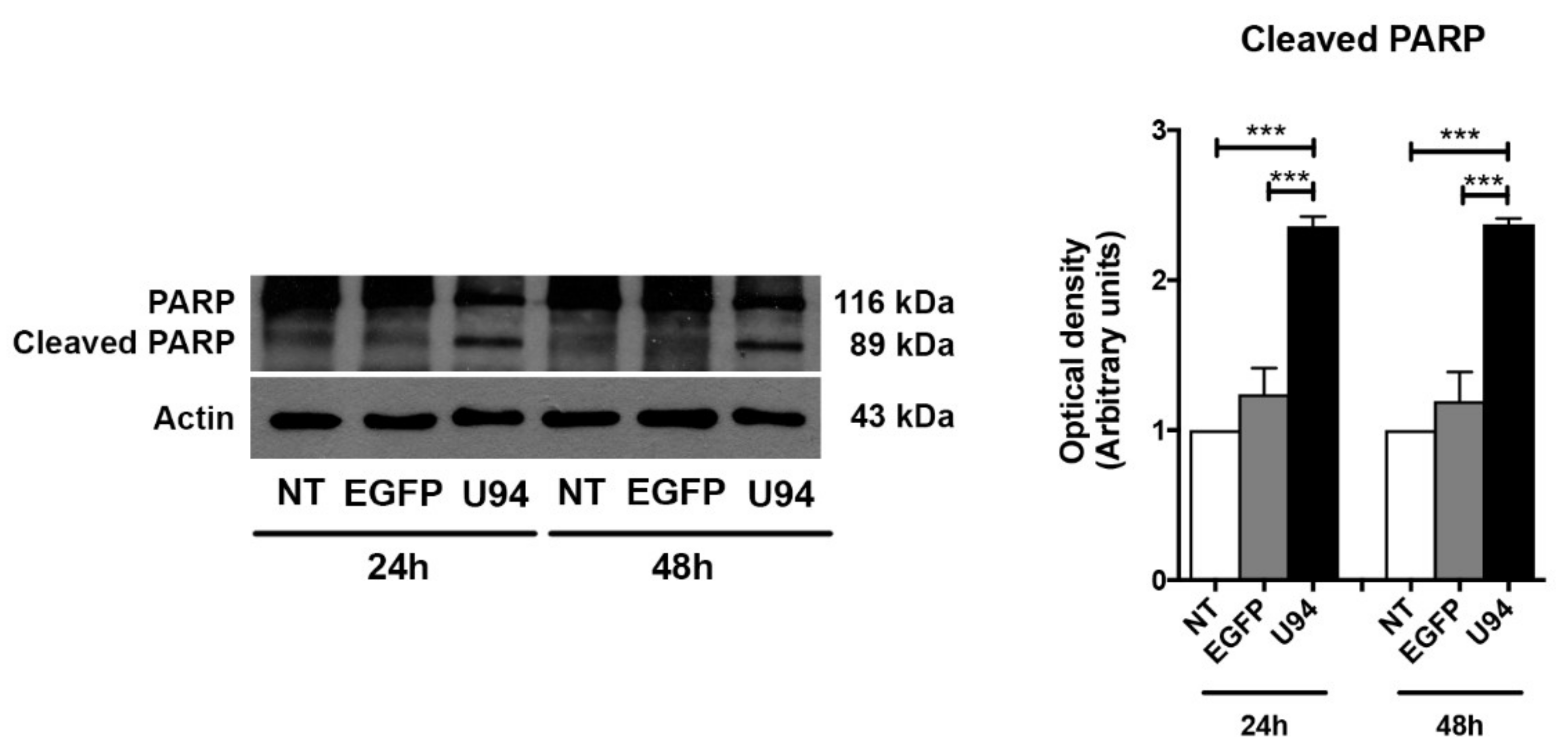
2.4. Activation of the Intrinsic Apoptotic Pathway by U94 Expression
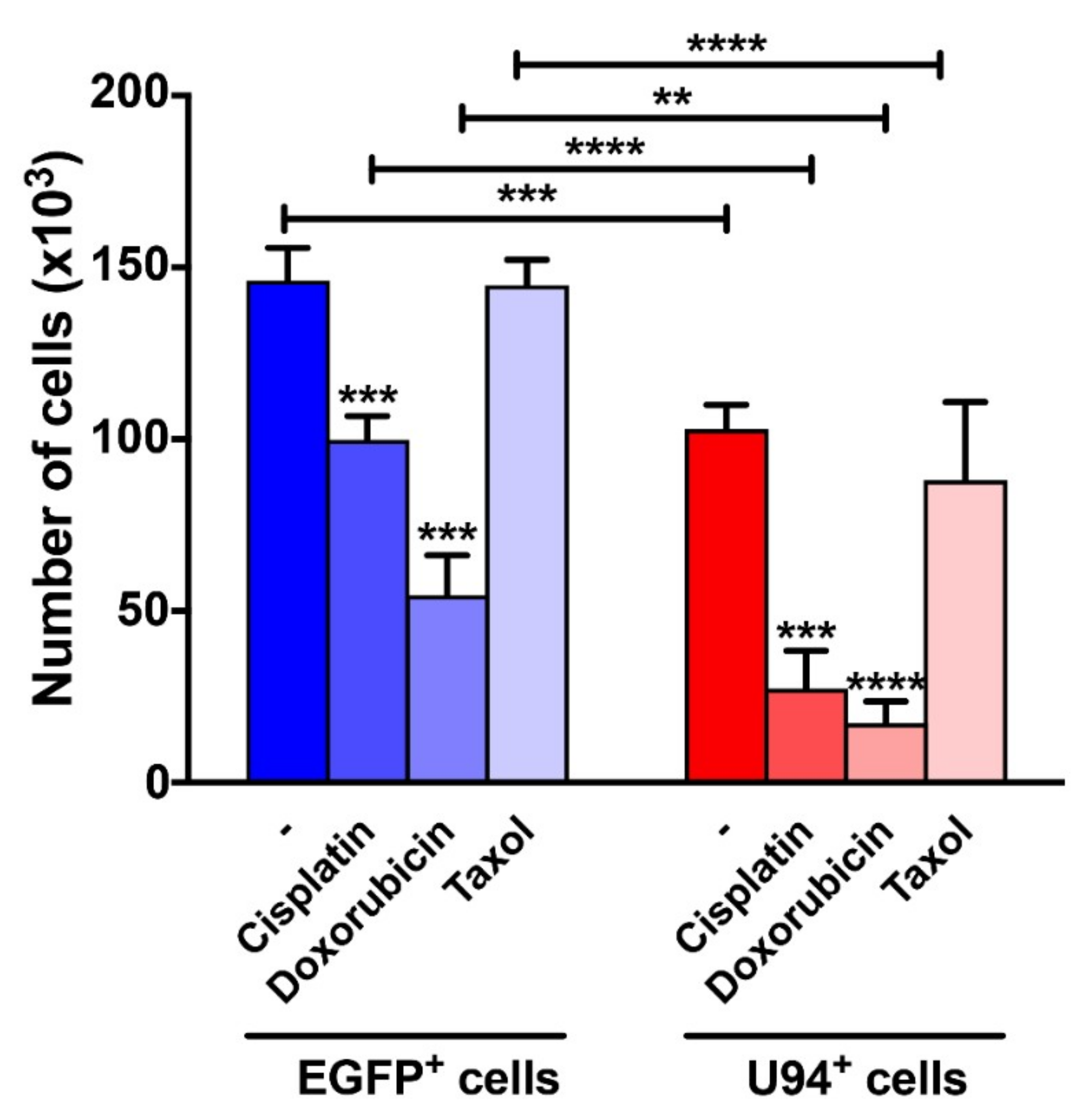
2.5. Synergism of U94 and DNA-Damaging Drugs against Breast Cancer Cells
2.6. U94 Induces Apoptosis in Different TNBCs
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Cell Transduction
4.3. Reagents
4.4. Gene Expression Profile and Bioinformatic Analysis
4.5. Real-Time PCR
4.6. Annexin V Assay
4.7. Western Blot Analysis
4.8. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
4.9. Chemotherapeutic Drugs Cytotoxic Activity on MDA-MB 231 Cell Line Upon U94 Expression
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ginsburg, O.; Bray, F.; Coleman, M.P.; Vanderpuye, V.; Eniu, A.; Kotha, S.R.; Sarker, M.; Huong, T.T.; Allemani, C.; Dvaladze, A.; et al. The global burden of women’s cancers: A grand challenge in global health. Lancet 2017, 389, 847–860. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, G.; Rajala, M.S. Combination of Oncolytic Measles Virus Armed With BNiP3, a Pro-apoptotic Gene and Paclitaxel Induces Breast Cancer Cell Death. Front. Oncol. 2019, 8, 676. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2017, 8, 1913–1924. [Google Scholar] [CrossRef] [PubMed]
- Sporikova, Z.; Koudelakova, V.; Trojanec, R.; Hajduch, M. Genetic Markers in Triple-Negative Breast Cancer. Clin. Breast Cancer 2018, 18, e841–e850. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Heitz, F.; Harter, P.; Lueck, H.J.; Fissler-Eckhoff, A.; Lorenz-Salehi, F.; Scheil-Bertram, S.; Traut, A.; du Bois, A. Triple-negative and HER2-overexpressing breast cancers exhibit an elevated risk and an earlier occurrence of cerebral metastases. Eur. J. Cancer. 2009, 45, 2792–2798. [Google Scholar] [CrossRef]
- Liedtke, C.; Hess, K.R.; Karn, T.; Rody, A.; Kiesel, L.; Hortobagyi, G.N.; Pusztai, L.; Gonzalez-Angulo, A.M. The prognostic impact of age in patients with triple-negative breast cancer. Breast Cancer Res. Treat. 2013, 138, 591–599. [Google Scholar] [CrossRef]
- Yuan, N.; Meng, M.; Liu, C.; Feng, L.; Hou, L.; Ning, Q.; Xin, G.; Pei, L.; Gu, S.; Li, X.; et al. Clinical characteristics and prognostic analysis of triple-negative breast cancer patients. Mol. Clin. Oncol. 2014, 2, 245–251. [Google Scholar] [CrossRef]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, S142–S150. [Google Scholar] [CrossRef] [PubMed]
- Berrada, N.; Delaloge, S.; André, F. Treatment of triple-negative metastatic breast cancer: Toward individualized targeted treatments or chemosensitization? Ann. Oncol. 2010, 21, vii30–vii35. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Alemany, R. Viruses in cancer treatment. Clin. Transl. Oncol. 2013, 15, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Caruso, A.; Caselli, E.; Fiorentini, S.; Rotola, A.; Prandini, A.; Garrafa, E.; Saba, E.; Alessandri, G.; Cassai, E.; Di Luca, D. U94 of human herpesvirus 6 inhibits in vitro angiogenesis and lymphangiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 20446–20451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caselli, E.; Bracci, A.; Galvan, M.; Boni, M.; Rotola, A.; Bergamini, C.; Cermelli, C.; Dal Monte, P.; Gompels, U.A.; Cassai, E.; et al. Human herpesvirus 6 (HHV-6) U94/REP protein inhibits betaherpesvirus replication. Virology 2006, 346, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Dhepakson, P.; Mori, Y.; Jiang, Y.B.; Huang, H.L.; Akkapaiboon, P.; Okuno, T.; Yamanishi, K. Human herpesvirus-6 rep/U94 gene product has single-stranded DNA-binding activity. J. Gen. Virol. 2002, 83, 847–854. [Google Scholar] [CrossRef]
- Mori, Y.; Dhepakson, P.; Shimamoto, T.; Ueda, K.; Gomi, Y.; Tani, H.; Matsuura, Y.; Yamanishi, K. Expression of human herpesvirus 6B rep within infected cells and binding of its gene product to the TATA-binding protein in vitro and in vivo. J. Virol. 2000, 74, 6096–6104. [Google Scholar] [CrossRef]
- Trempe, F.; Gravel, A.; Dubuc, I.; Wallaschek, N.; Collin, V.; Gilbert-Girard, S.; Morissette, G.; Kaufer, B.B.; Flamand, L. Characterization of human herpesvirus 6A/B U94 asnATPase, helicase, exonuclease and DNA-binding proteins. Nucleic Acids Res. 2015, 43, 6084–6098. [Google Scholar] [CrossRef]
- Araujo, J.C.; Doniger, J.; Stöppler, H.; Sadaie, M.R.; Rosenthal, L.J. Cell lines containing and expressing the human herpesvirus 6A ts gene are protected from both H-ras and BPV-1 transformation. Oncogene 1997, 14, 937–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ifon, E.T.; Pang, A.L.; Johnson, W.; Cashman, K.; Zimmerman, S.; Muralidhar, S.; Chan, W.Y.; Casey, J.; Rosenthal, L.J. U94 alters FN1 and ANGPTL4 gene expression and inhibits tumorigenesis of prostate cancer cell line PC3. Cancer Cell Int. 2005, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Caccuri, F.; Ronca, R.; Laimbacher, A.S.; Berenzi, A.; Steimberg, N.; Campilongo, F.; Mazzuca, P.; Giacomini, A.; Mazzoleni, G.; Benetti, A.; et al. U94 of human herpesvirus 6 down-modulates Src, promotes a partial mesenchymal-to-epithelial transition and inhibits tumor cell growth, invasion and metastasis. Oncotarget 2017, 8, 44533–44549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, B.; Li, L.; Li, M.; Wang, J.; Zhang, G.; Yao, K.; Wang, S. U94/rep of human herpesvirus 6 inhibits proliferation, invasion, and angiogenesis of glioma. Cancer Manag. Res. 2018, 10, 5991–6001. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, L.; Tang, H.; Gu, B.; Feng, D.; Zhou, F.; Liu, Y.; Xu, X.; Chen, Y.; Yao, K.; et al. Human herpesvirus 6 U94 suppresses tumor cell proliferation and invasion by inhibiting Akt/GSK3β signaling in glioma. Transl. Cancer Res. 2018, 7, 1694–1702. [Google Scholar] [CrossRef]
- Bergkvist, A.; Rusnakova, V.; Sindelka, R.; Garda, J.M.; Sjögreen, B.; Lindh, D.; Forootan, A.; Kubista, M. Gene expression profiling—Clusters of possibilities. Methods 2010, 50, 323–335. [Google Scholar] [CrossRef]
- Hung, J.H.; Yang, T.H.; Hu, Z.; Weng, Z.; DeLisi, C. Gene set enrichment analysis: Performance evaluation and usage guidelines. Brief. Bioinform. 2012, 13, 281–291. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Yu, C.; Cao, H.; He, X.; Sun, P.; Feng, Y.; Chen, L.; Gong, H. Cyclin-dependent kinase inhibitor 3 (CDKN3) plays a critical role in prostate cancer via regulating cell cycle and DNA replication signaling. Biomed. Pharmacother. 2017, 96, 1109–1118. [Google Scholar] [CrossRef]
- Deng, M.; Wang, J.; Chen, Y.; Zhang, L.; Xie, G.; Liu, Q.; Zhang, T.; Yuan, P.; Liu, D. Silencing cyclin-dependent kinase inhibitor 3 inhibits the migration of breast cancer cell lines. Mol. Med. Rep. 2016, 14, 1523–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, P.; Xi, G.M.; Zhang, B.; Wu, Y.; Liu, H.B.; Liu, Y.F.; Xu, W.J.; Zhu, Q.; Cai, F.; Zhou, Z.J.; et al. NCAPG2 promotes tumour proliferation by regulating G2/M phase and associates with poor prognosis in lung adenocarcinoma. J. Cell. Mol. Med. 2017, 21, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Chen, X.; Sun, J.; Bie, P.; Zhang, L.D. siRNA-mediated knockdown against NUF2 suppresses pancreatic cancer proliferation in vitro and in vivo. Biosci. Rep. 2015, 35, e00170. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.L.; Shao, L. Silencing of NUF2 inhibits proliferation of human osteosarcoma Saos-2 cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 1071–1079. [Google Scholar] [PubMed]
- Hoeijmakers, J.H.; Bootsma, D. DNA repair: Incisions for excision. Nature 1994, 371, 654–655. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.; Zhou, F.; Yang, H.; Wei, Y.; Gong, J.; Mei, Z.; Wu, L.; Yu, H.; Zhou, Y. Downregulation of high mobility group box 1 modulates telomere homeostasis and increases the radiosensitivity of human breast cancer cells. Int. J. Oncol. 2015, 46, 1051–1058. [Google Scholar] [CrossRef]
- He, S.J.; Cheng, J.; Feng, X.; Yu, Y.; Tian, L.; Huang, Q. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget 2017, 8, 64534–64550. [Google Scholar] [CrossRef] [Green Version]
- Lange, S.S.; Mitchell, D.L.; Vasquez, K.M. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 10320–10325. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.H.; Xu, M. DNA fragmentation in apoptosis. Cell Res. 2000, 10, 205–211. [Google Scholar] [CrossRef]
- Eidet, J.R.; Pasovic, L.; Maria, R.; Jackson, C.J.; Utheim, T.P. Objective assessment of changes in nuclear morphology and cell distribution following induction of apoptosis. Diagn. Pathol. 2014, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Pietkiewicz, S.; Schmidt, J.H.; Lavrik, I.N. Quantification of apoptosis and necroptosis at the single cell level by a combination of Imaging Flow Cytometry with classical Annexin V/propidium iodide staining. J. Immunol. Methods 2015, 423, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Kutuk, O.; Brito, G.C.; Andrews, T.S.; Leber, B.; Letai, A.; Andrews, D.W. Phosphorylation switches Bax from promoting to inhibiting apoptosis thereby increasing drug resistance. EMBO Rep. 2018, 19, e45235. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Bouchon, A.; Stahl, H.; Krammer, P.H. Tumor necrosis factor-related apoptosis-inducing ligand retains its apoptosis-inducing capacity on Bcl-2- or Bcl-xL-overexpressing chemotherapy-resistant tumor cells. Cancer Res. 2000, 60, 3051–3057. [Google Scholar] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Yang, E.S. The intersection between DNA damage response and cell death pathways. Exp. Oncol. 2012, 34, 243–254. [Google Scholar] [PubMed]
- DeBiasi, R.L.; Clarke, P.; Meintzer, S.; Jotte, R.; Kleinschmidt-Demasters, B.K.; Johnson, G.L.; Tyler, K.L. Reovirus-induced alteration in expression of apoptosis and DNA repair genes with potential roles in viral pathogenesis. J. Virol. 2003, 77, 8934–8947. [Google Scholar] [CrossRef]
- Fox, C.R.; Parks, G.D. Parainfluenza Virus Infection Sensitizes Cancer Cells to DNA-Damaging Agents: Implications for Oncolytic Virus Therapy. J. Virol. 2018, 92, e01948-17. [Google Scholar] [CrossRef] [PubMed]
- Egger, S.J.; Willson, M.L.; Morgan, J.; Walker, H.S.; Carrick, S.; Ghersi, D.; Wilcken, N. Platinum-containing regimens for metastatic breast cancer. Cochrane Database Syst. Rev. 2017, 6, CD003374. [Google Scholar] [CrossRef] [PubMed]
- Jasra, S.; Anampa, J. Anthracycline Use for Early Stage Breast Cancer in the Modern Era: A Review. Curr. Treat. Options Oncol. 2018, 19, 30. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Helleday, T. DNA REPAIR. Drugging DNA repair. Science 2016, 352, 1178–1179. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY). 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W. Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer 2005, 5, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.T.; Wu, C.Y.; Chung, C.Y.; Hwu, Y.; Cheng, S.H.; Mou, C.Y.; Lo, L.W. Probing the dynamics of doxorubicin-DNA intercalation during the initial activation of apoptosis by fluorescence lifetime imaging microscopy (FLIM). PLoS ONE 2012, 7, e44947. [Google Scholar]
- Jia, S.; Wang, R.; Wu, K.; Jiang, H.; Du, Z. Elucidation of the Mechanism of Action for Metal Based Anticancer Drugs by Mass Spectrometry-Based Quantitative Proteomics. Molecules 2019, 24, 581. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Bartek, J.; Bartkova, J.; Lukas, J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007, 26, 7773–7779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Buchwald, H. Cholesterol inhibition, cancer, and chemotherapy. Lancet 1992, 339, 1154–1156. [Google Scholar] [CrossRef]
- Cruz, P.M.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; D’Accolti, M.; Bortolotti, D.; Caccuri, F.; Caruso, A.; Di Luca, D.; Caselli, E. Human Herpesvirus 6A and 6B inhibit in vitro angiogenesis by induction of Human Leukocyte Antigen G. Sci. Rep. 2018, 8, 17683. [Google Scholar] [CrossRef]
- Miller, J.A.; Cai, C.; Langfelder, P.; Geschwind, D.H.; Kurian, S.M.; Salomon, D.R. Horvath S. Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinformatics 2011, 12, 322. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Le Noci, V.; Sommariva, M.; Tortoreto, M.; Zaffaroni, N.; Campiglio, M.; Tagliabue, E.; Balsari, A.; Sfondrini, L. Reprogramming the lung microenvironment by inhaled immunotherapy fosters immune destruction of tumor. Oncoimmunology. Oncoimmunology 2016, 5, e1234571. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caccuri, F.; Sommariva, M.; Marsico, S.; Giordano, F.; Zani, A.; Giacomini, A.; Fraefel, C.; Balsari, A.; Caruso, A. Inhibition of DNA Repair Mechanisms and Induction of Apoptosis in Triple Negative Breast Cancer Cells Expressing the Human Herpesvirus 6 U94. Cancers 2019, 11, 1006. https://doi.org/10.3390/cancers11071006
Caccuri F, Sommariva M, Marsico S, Giordano F, Zani A, Giacomini A, Fraefel C, Balsari A, Caruso A. Inhibition of DNA Repair Mechanisms and Induction of Apoptosis in Triple Negative Breast Cancer Cells Expressing the Human Herpesvirus 6 U94. Cancers. 2019; 11(7):1006. https://doi.org/10.3390/cancers11071006
Chicago/Turabian StyleCaccuri, Francesca, Michele Sommariva, Stefania Marsico, Francesca Giordano, Alberto Zani, Arianna Giacomini, Cornel Fraefel, Andrea Balsari, and Arnaldo Caruso. 2019. "Inhibition of DNA Repair Mechanisms and Induction of Apoptosis in Triple Negative Breast Cancer Cells Expressing the Human Herpesvirus 6 U94" Cancers 11, no. 7: 1006. https://doi.org/10.3390/cancers11071006
APA StyleCaccuri, F., Sommariva, M., Marsico, S., Giordano, F., Zani, A., Giacomini, A., Fraefel, C., Balsari, A., & Caruso, A. (2019). Inhibition of DNA Repair Mechanisms and Induction of Apoptosis in Triple Negative Breast Cancer Cells Expressing the Human Herpesvirus 6 U94. Cancers, 11(7), 1006. https://doi.org/10.3390/cancers11071006