Inhibition of Protein Phosphatase 1 Stimulates Noncanonical ER Stress eIF2α Activation to Enhance Fisetin-induced Chemosensitivity in HDAC Inhibitor-resistant Hepatocellular Carcinoma Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Influence of Fisetin on Normal and Liver-Cancer Cells
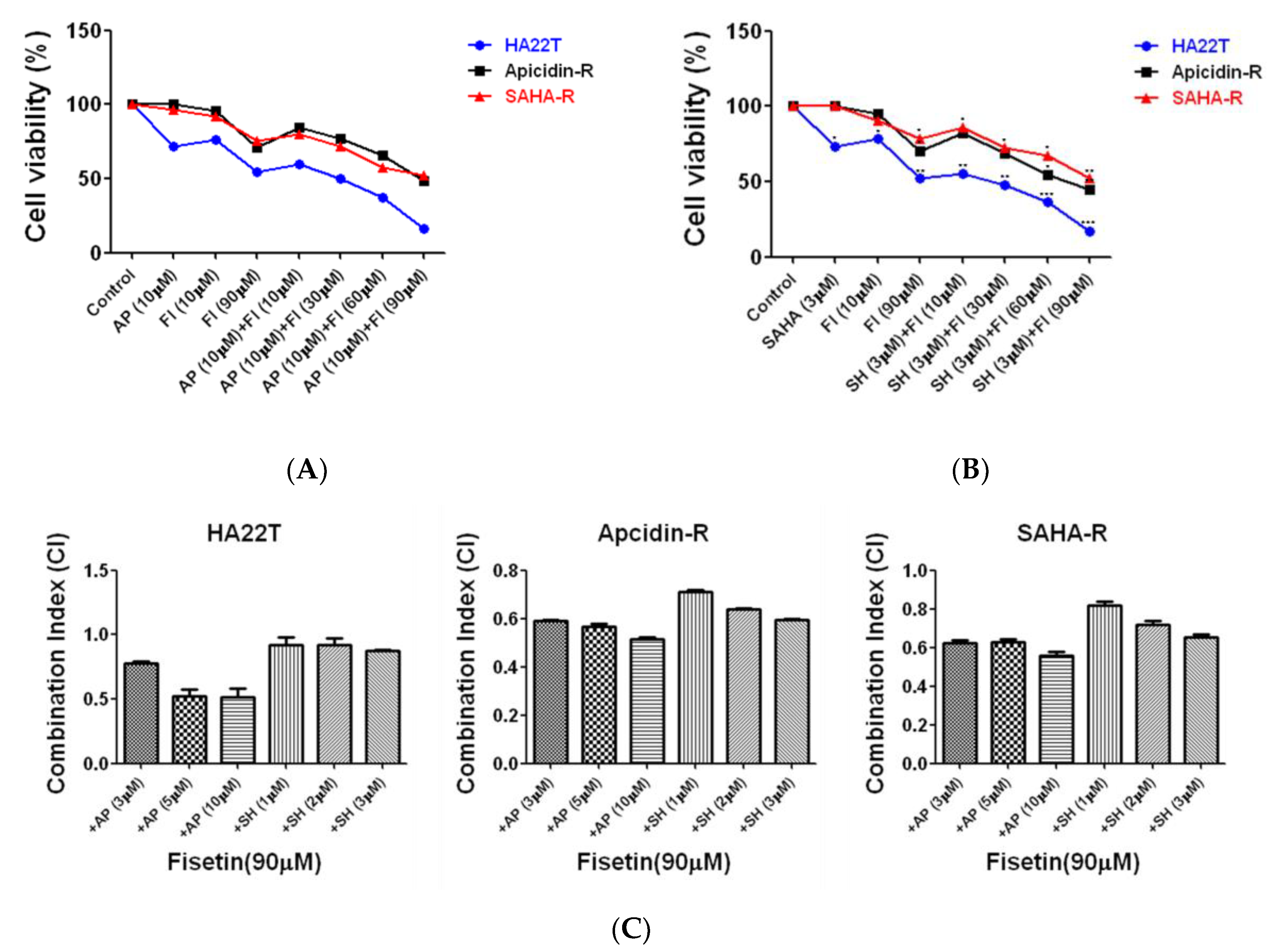
2.2. Assessment of Fisetin as a Complementary Therapy in Hepatocellular Carcinoma Chemotherapy
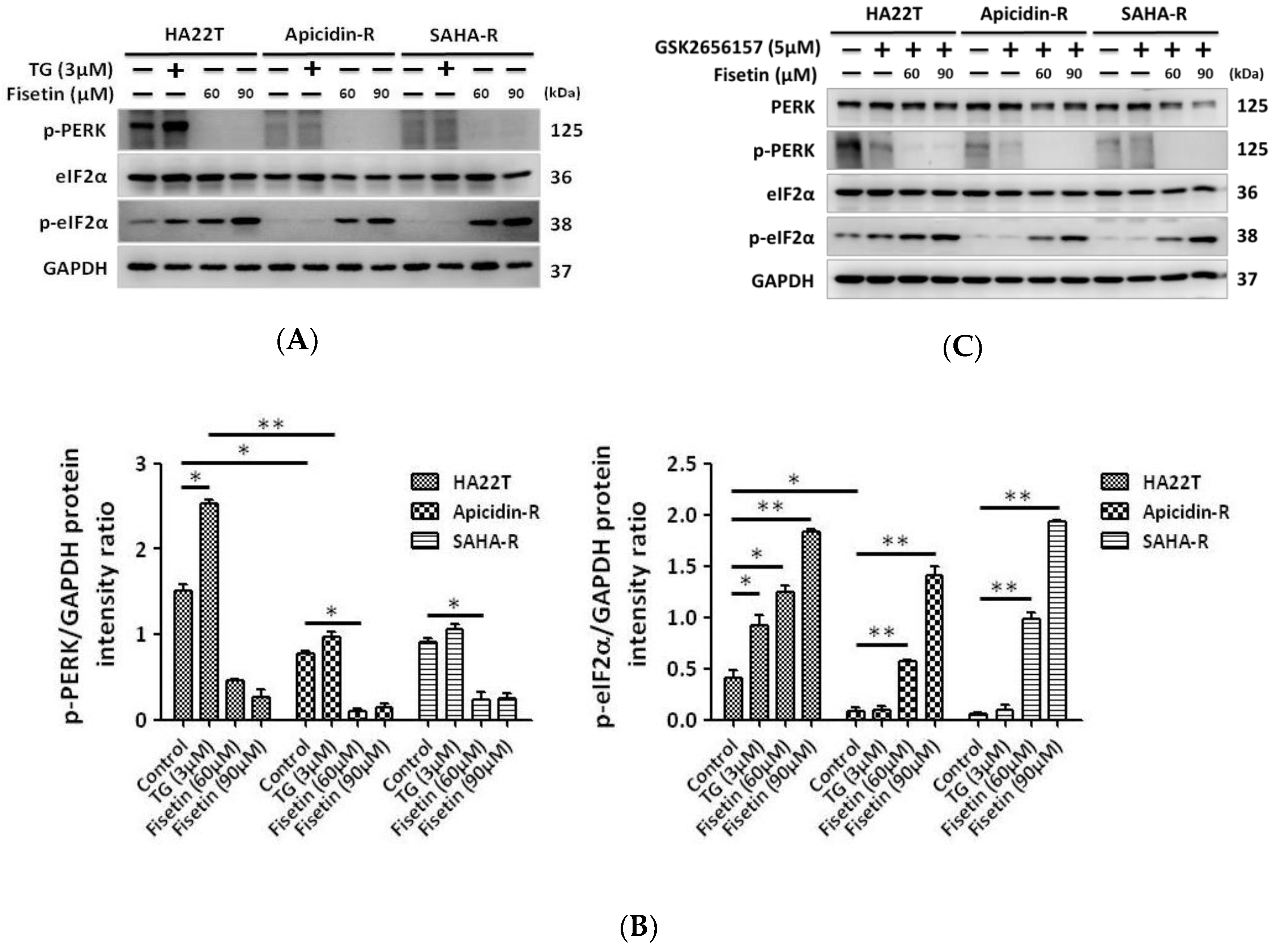
2.3. The ER Stress-Dependent Pathway Was not Significantly Involved in the Effect Of Fisetin on Liver Cancer Cells
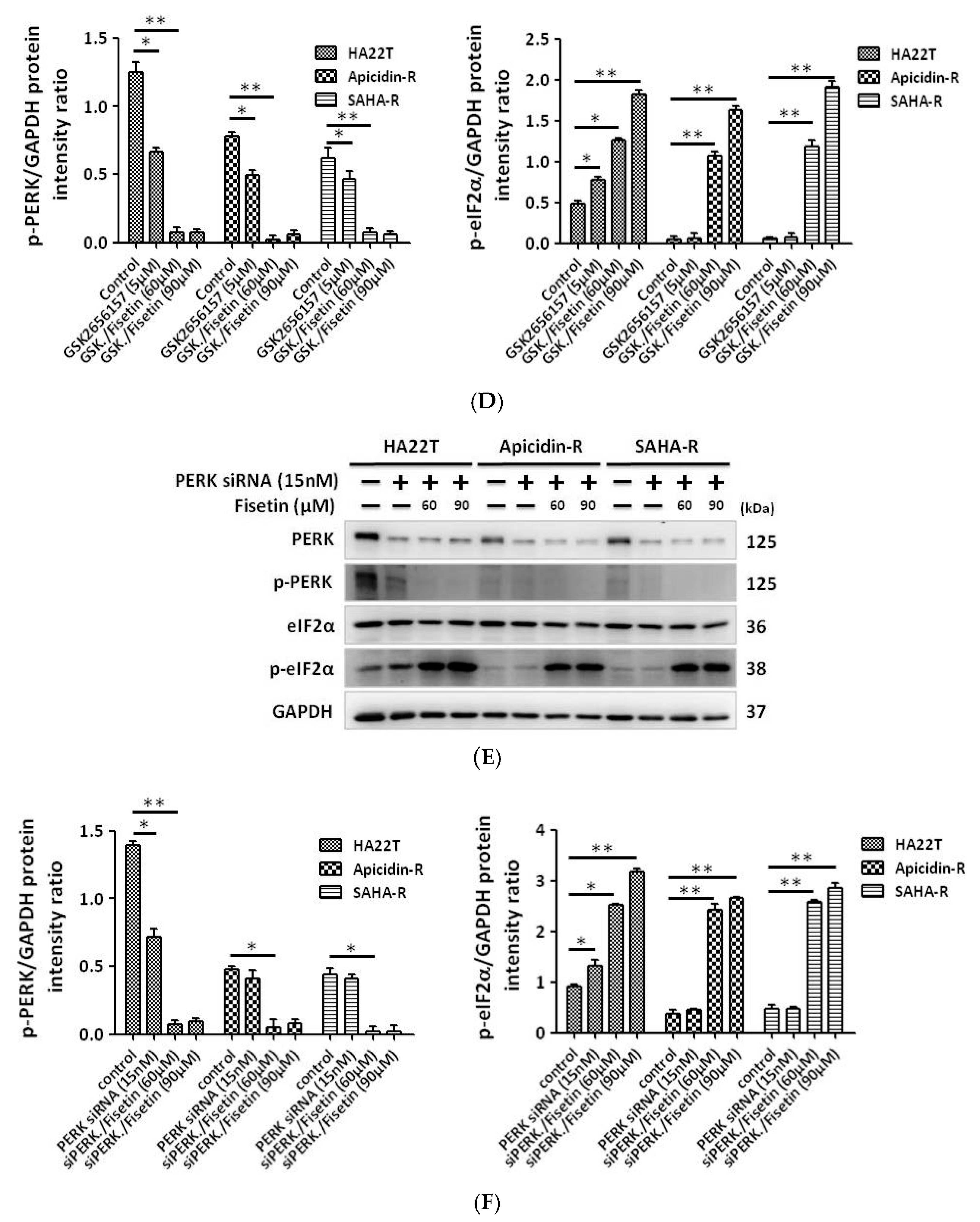
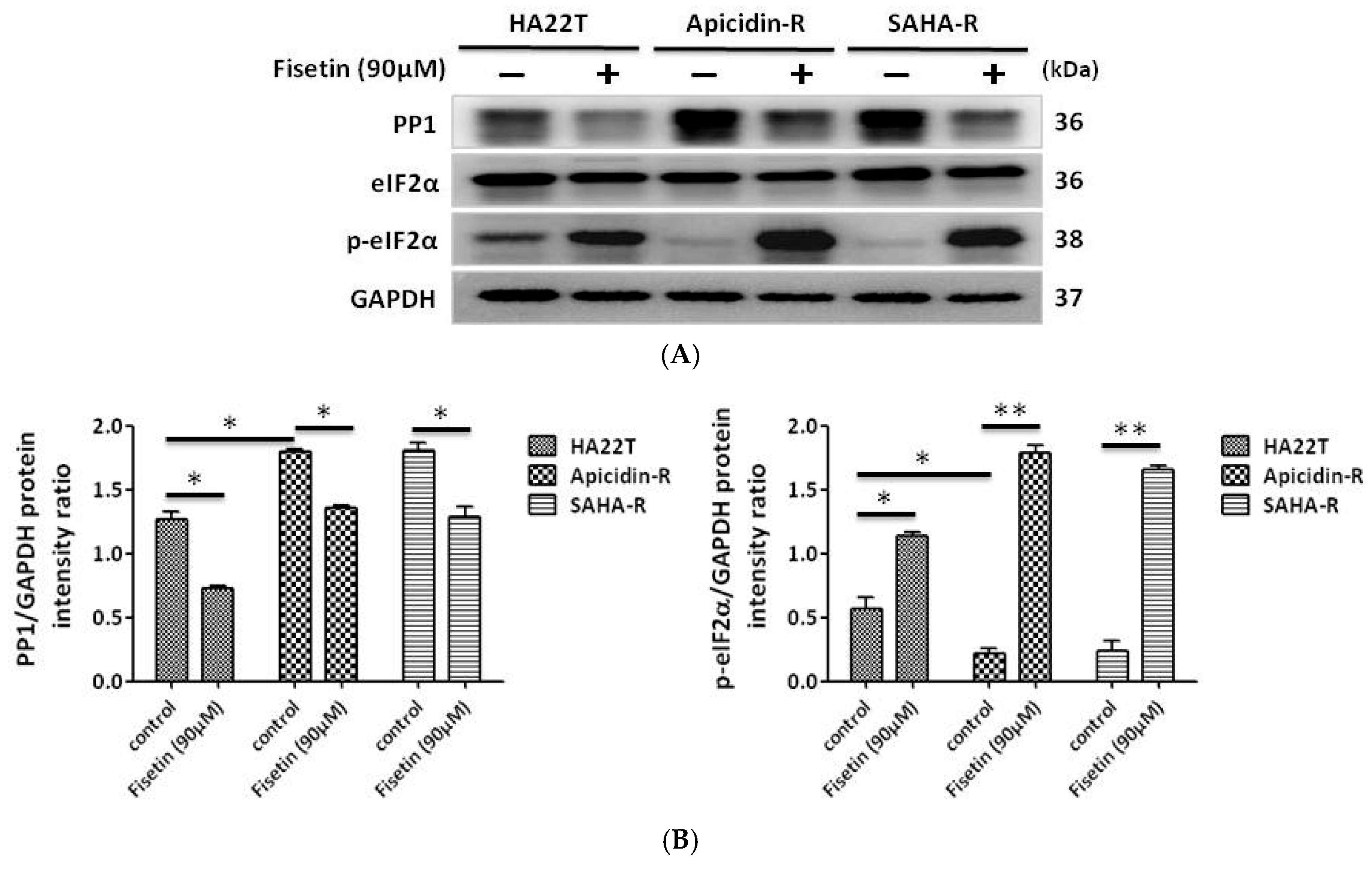
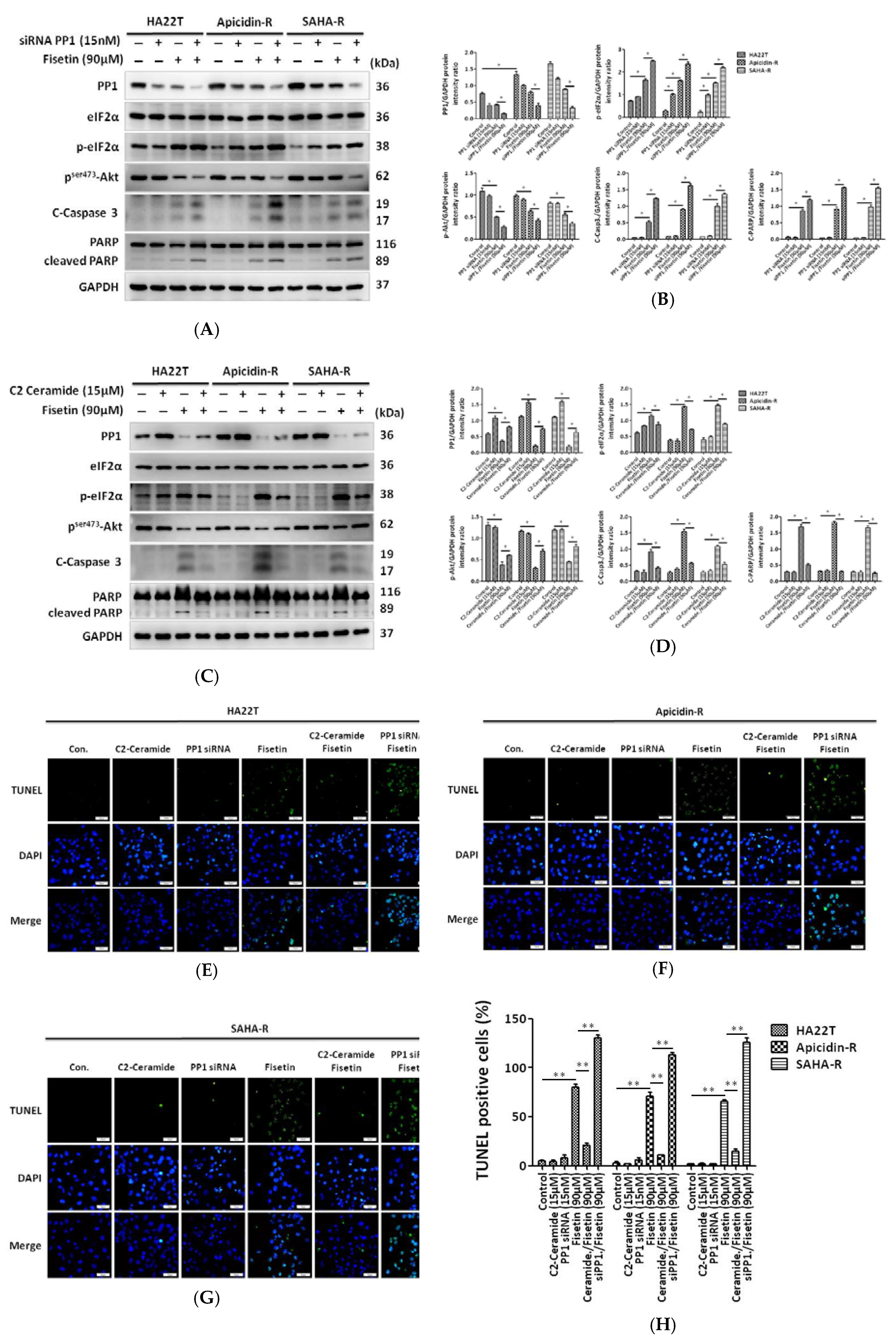
2.4. Fisetin Induced the Phosphorylation of eIF-2α via the Inhibition of PP1 Expression in HCC Cells
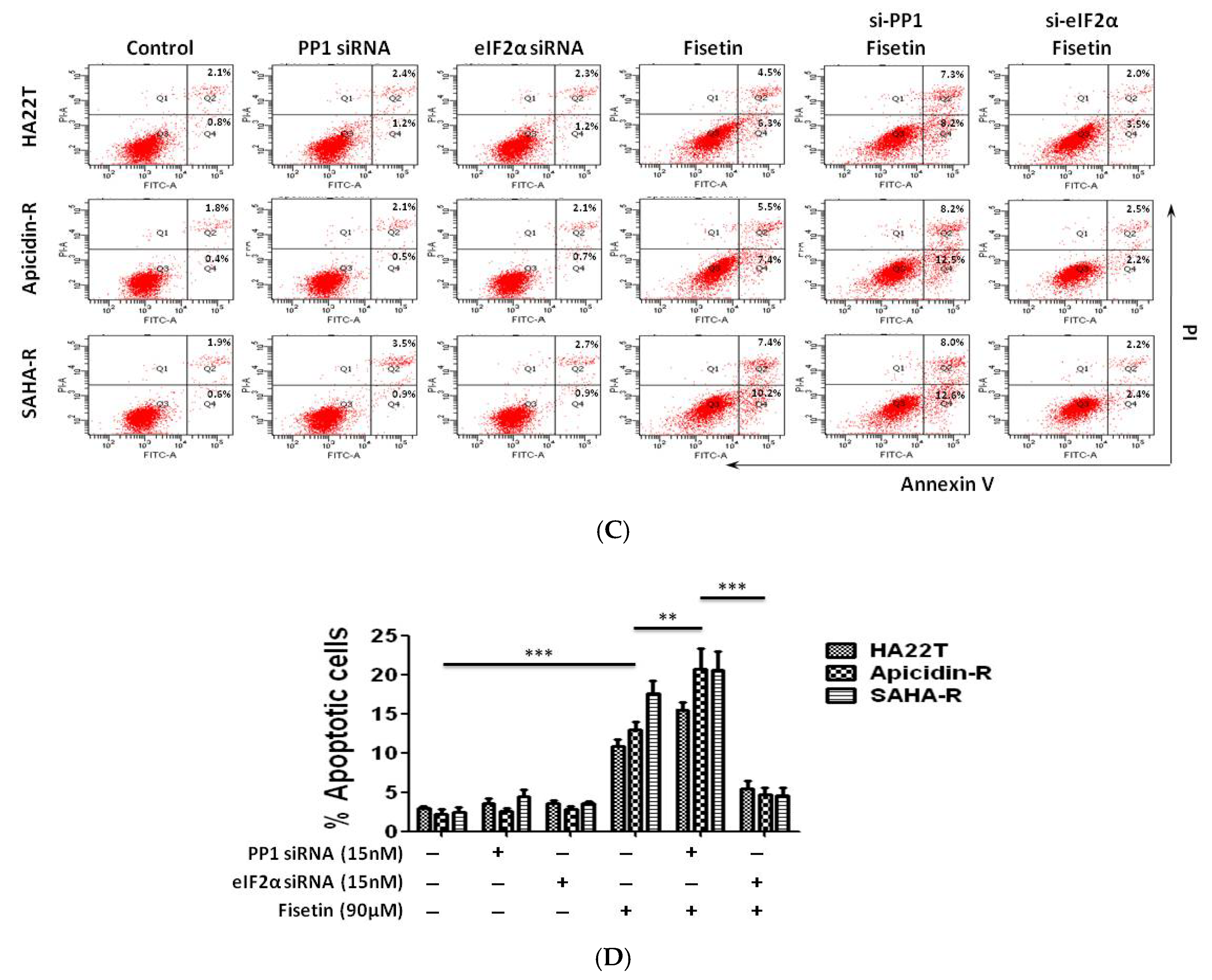
2.5. Protein Phosphatase 1 Is Significantly Engaged in the Effect of Fisetin on HCC Apoptosis
2.6. Antidrug Effect of p-eIF2α on HDACis-R and Parental Cell Lines
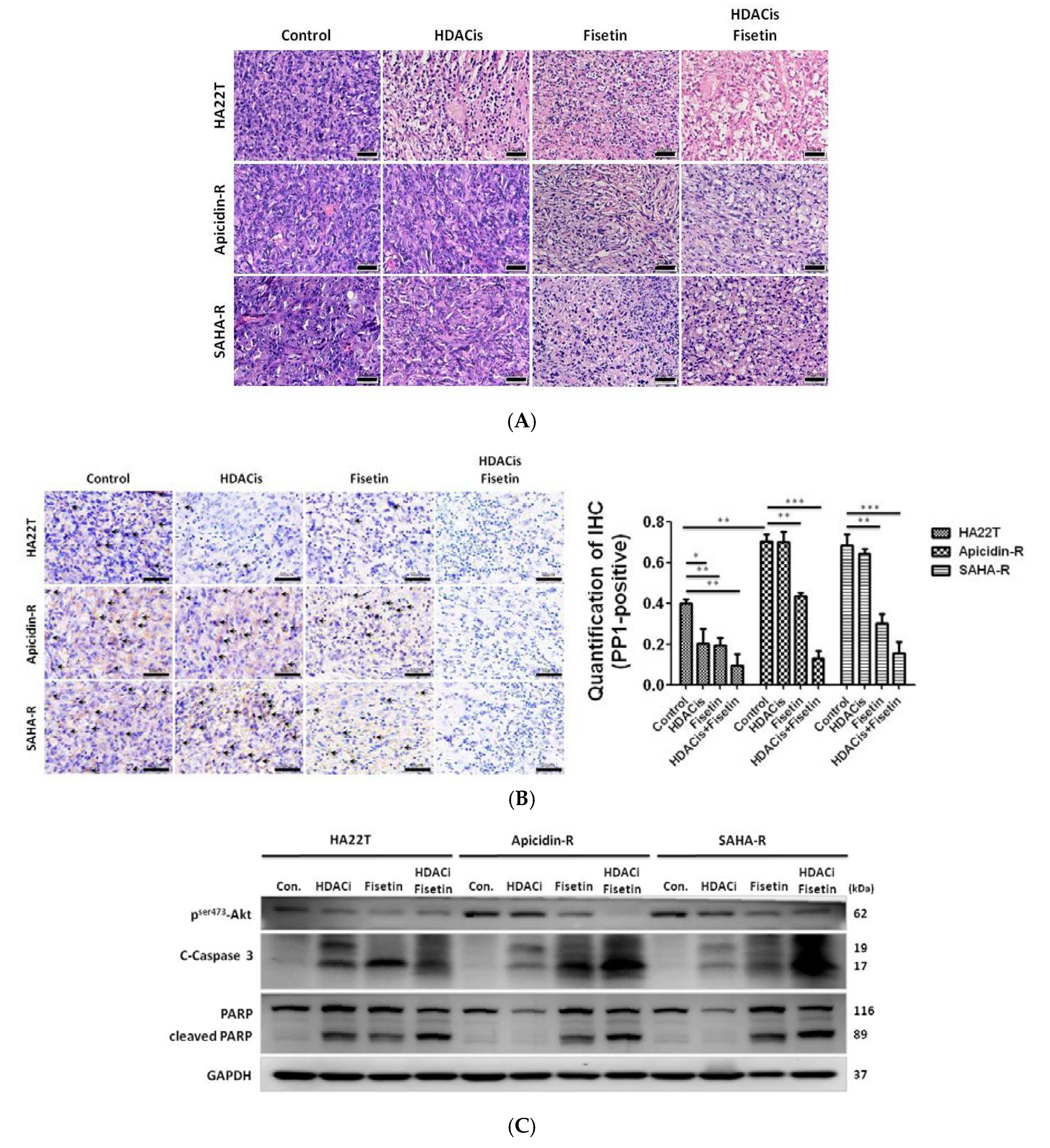
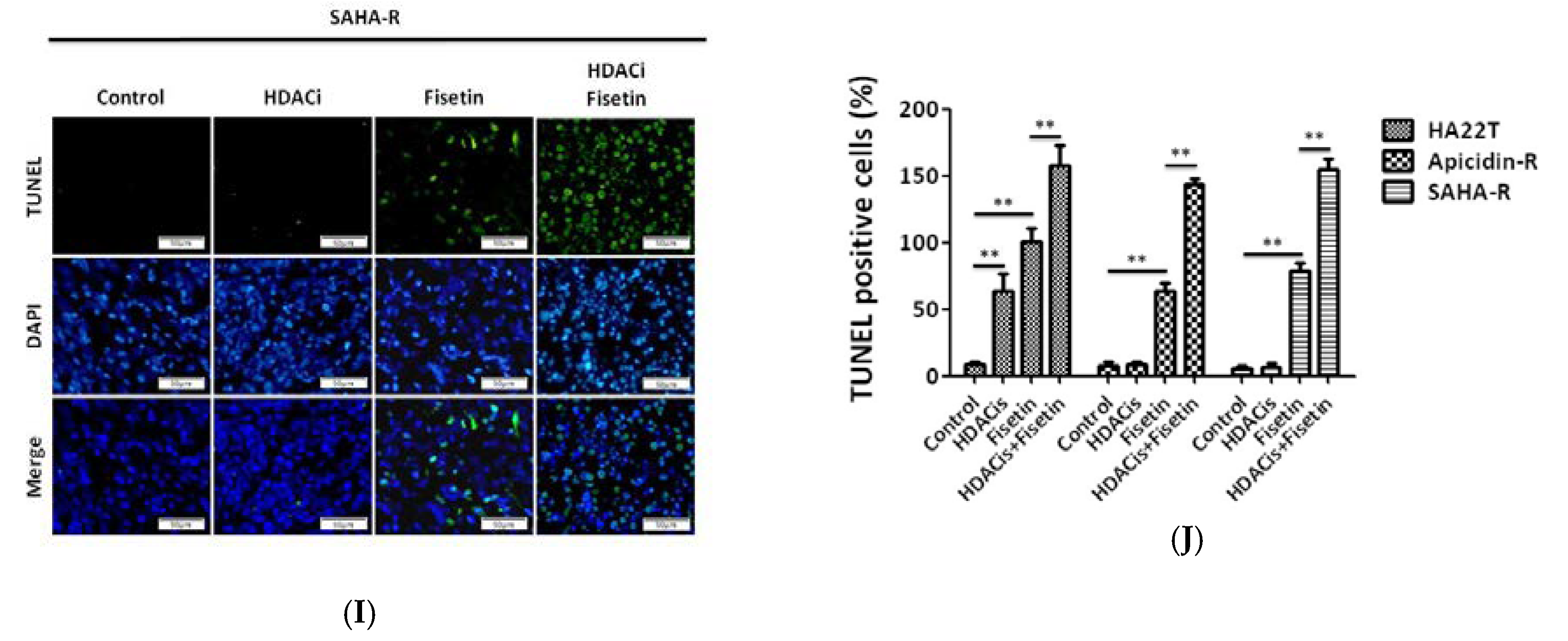
2.7. Fisetin Enhanced the Therapeutic Potential in Xenograft Tumors Generated from HDAC Inhibitor-Resistant Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Establishment of Resistant Cells
4.2. Whole-Cell Extraction
4.3. Cell Viability Assay
4.4. Antibodies and Drug formulations
4.5. Western Blot Analysis
4.6. TUNEL assay and DAPI Staining
4.7. FITC-Annexin V Staining for Apoptosis
4.8. Animal Model
4.9. Histopathological Examination
4.10. Immunohistochemistry
4.11. Statistical Analysis
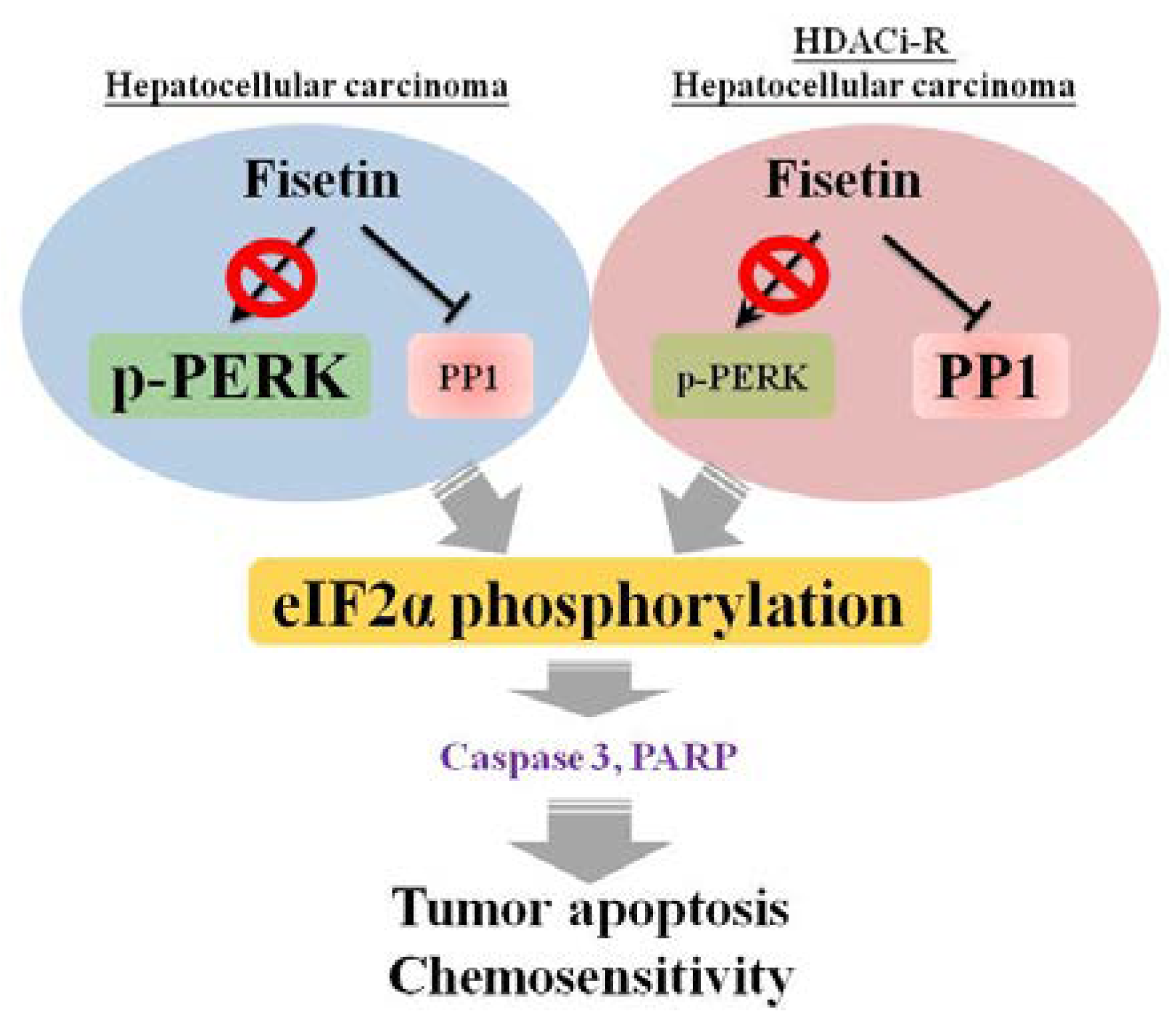
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, W.S.-W.; Lin, H.-Y.; Yeh, C.-B.; Chen, L.-Y.; Chou, Y.-E.; Yang, S.-F.; Liu, Y.-F.J.I.j.o.m.s. Correlation of chitinase 3-like 1 single nucleotide polymorphisms with hepatocellular carcinoma in Taiwan. Int. J. Med. Sci. 2017, 14, 136. [Google Scholar] [CrossRef] [PubMed]
- Serper, M.; Taddei, T.H.; Mehta, R.; D’Addeo, K.; Dai, F.; Aytaman, A.; Baytarian, M.; Fox, R.; Hunt, K.; Goldberg, D.S.J.G. Association of provider specialty and multidisciplinary care with hepatocellular carcinoma treatment and mortality. Gastroenterology 2017, 152, 1954–1964. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.; Carter, A.; Casey, D.; Charlson, F.; Chen, A.; Coates, M.; Coggeshall, M. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef]
- Starzl, T.E.; Marchioro, T.; Von Kaulla, K.; Hermann, G.; Brittain, R.; Waddell, W. Homotransplantation of the liver in humans. Surg. Gynecol. Obstet. 1963, 117, 659. [Google Scholar]
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Vinals, F.; Capella, G. Recent advances in cancer therapy: an overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Song, B.; Sun, G.; Ma, T.; Zhong, F.; Wei, W. Endoplasmic Reticulum Stress–Induced Resistance to Doxorubicin Is Reversed by Paeonol Treatment in Human Hepatocellular Carcinoma Cells. PloS One 2013, 8, e62627. [Google Scholar] [CrossRef]
- Wickremasinghe, R.G.; Hoffbrand, A.V. Biochemical and genetic control of apoptosis: relevance to normal hematopoiesis and hematological malignancies. Blood 1999, 93, 3587–3600. [Google Scholar]
- Cheng, X.; Bennett, R.; Liu, X.; Byrne, M.; May, W.S. PKR negatively regulates leukemia progression in association with PP2A activation, Bcl-2 inhibition and increased apoptosis. Blood Cancer J. 2013, 3, e144. [Google Scholar] [CrossRef]
- Steelman, L.; Franklin, R.; Abrams, S.; Chappell, W.; Kempf, C.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.J.L. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080. [Google Scholar] [CrossRef]
- Hsu, H.-H.; Cheng, L.-H.; Ho, T.-J.; Kuo, W.-W.; Lin, Y.-M.; Chen, M.-C.; Lee, N.-H.; Tsai, F.-J.; Tsai, K.-H.; Huang, C.-Y.J.T.B. Apicidin-resistant HA22T hepatocellular carcinoma cells massively promote pro-survival capability via IGF-IR/PI3K/Akt signaling pathway activation. Tumor Biol. 2014, 35, 303–313. [Google Scholar] [CrossRef]
- Shi, Y. Serine/threonine phosphatases: mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Gong, L.; Yuan, D.; Deng, M.; Zeng, X.; Chen, L.; Zhang, L.; Yan, Q.; Liu, J.; Hu, X. Protein phosphatase-1 regulates Akt1 signal transduction pathway to control gene expression, cell survival and differentiation. Cell Death Differ. 2010, 17, 1448. [Google Scholar] [CrossRef] [PubMed]
- Li, D.W.; Liu, J.; Schmid, P.; Schlosser, R.; Feng, H.; Liu, W.; Yan, Q.; Gong, L.; Sun, S.; Deng, M. Protein serine/threonine phosphatase-1 dephosphorylates p53 at Ser-15 and Ser-37 to modulate its transcriptional and apoptotic activities. Oncogene 2006, 25, 3006. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-H.; Liu, C.W.; Avramis, V.I.; Berndt, N. Protein phosphatase 1α-mediated stimulation of apoptosis is associated with dephosphorylation of the retinoblastoma protein. Oncogene 2001, 20, 6111. [Google Scholar] [CrossRef] [PubMed]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-Y.; Wang, L.-T.; Hsu, S.-H. Modification of epigenetic histone acetylation in hepatocellular carcinoma. Cancers 2018, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, M.; Sun, H.; Yin, K. Matrine improves cognitive impairment and modulates the balance of Th17/Treg cytokines in a rat model of Aβ1-42-induced Alzheimer's disease. 2015, 40, 411. Cent.-Eur. J. Immunol. 2015, 40, 411. [Google Scholar] [CrossRef] [PubMed]
- Tsilimigras, D.I.; Ntanasis-Stathopoulos, I.; Moris, D.; Spartalis, E.; Pawlik, T.M. Histone deacetylase inhibitors in hepatocellular carcinoma: A therapeutic perspective. Surgical Oncol. 2018. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Breslow, R.; Rifkind, R.A. Histone deacetylase inhibitors as new cancer drugs. Current Opin. Oncol. 2001, 13, 477–483. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Cheng, L.-H.; Hsu, H.-H.; Ho, T.-J.; Tu, C.-C.; Lin, Y.-M.; Chen, M.-C.; Tsai, F.-J.; Hsieh, Y.-L.; Huang, C.-Y.J.B. Apicidin-resistant HA22T hepatocellular carcinoma cells strongly activated the Wnt/β-catenin signaling pathway and MMP-2 expression via the IGF-IR/PI3K/Akt signaling pathway enhancing cell metastatic effect. Biosci. Biotechnol. Biochem. 2013, 77, 2397–2404. [Google Scholar]
- Tu, C.-C.; Cheng, L.-H.; Hsu, H.-H.; Chen, L.-M.; Lin, Y.-M.; Chen, M.-C.; Lee, N.-H.; Tsai, F.-J.; Huang, C.-Y.; Wu, W.-J.J.C.J.P. Activation of snail and EMT-like signaling via the IKKαβ/NF-κB pathway in Apicidin-resistant HA22T hepatocellular carcinoma cells. Chin. J. Physiol. 2013, 56, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Hsu, H.; Chen, T.; Chen, M.; Day, C.; Tu, C.; Lin, Y.; Tsai, F.; Kuo, W.; Huang, C.J.O. Phosphorylation of cofilin-1 by ERK confers HDAC inhibitor resistance in hepatocellular carcinoma cells via decreased ROS-mediated mitochondria injury. Oncogene 2017, 36, 1978. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.P.; Natarajan, L.; Caan, B.J.; Parker, B.A.; Greenberg, E.R.; Flatt, S.W.; Rock, C.L.; Kealey, S.; Al-Delaimy, W.K.; Bardwell, W.A. Influence of a diet very high in vegetables, fruit, and fiber and low in fat on prognosis following treatment for breast cancer: the Women's Healthy Eating and Living (WHEL) randomized trial. Jama 2007, 298, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Ju, D.-T.; Chang, C.-F.; Reddy, P.M.; Velmurugan, B.K.J.B. A review on the effects of current chemotherapy drugs and natural agents in treating non–small cell lung cancer. Biomedicine 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-H.; Chen, M.-C.; Day, C.H.; Lin, Y.-M.; Li, S.-Y.; Tu, C.-C.; Padma, V.V.; Shih, H.-N.; Kuo, W.-W.; Huang, C.-Y. Thymoquinone suppresses migration of LoVo human colon cancer cells by reducing prostaglandin E2 induced COX-2 activation. World J. Gastroenterol. 2017, 23, 1171. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.H.; Kuo, W.W.; Day, C.H.; Shibu, M.A.; Li, S.Y.; Chang, S.H.; Shih, H.N.; Chen, R.J.; Viswanadha, V.P.; Kuo, Y.H. Taiwanin E inhibits cell migration in human LoVo colon cancer cells by suppressing MMP-2/9 expression via p38 MAPK pathway. EnTox 2017, 32, 2021–2031. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Hsu, H.H.; Shibu, M.A.; Day, C.H.; Bau, D.T.; Ho, C.C.; Lin, Y.M.; Chen, M.C.; Wang, S.H.; Huang, C.Y. Down-regulation of β-catenin and the associated migration ability by Taiwanin C in arecoline and 4-NQO-induced oral cancer cells via GSK-3β activation. Mol. Carcinogenes. 2017, 56, 1055–1067. [Google Scholar] [CrossRef]
- Khan, N.; Afaq, F.; Syed, D.N.; Mukhtar, H. Fisetin, a novel dietary flavonoid, causes apoptosis and cell cycle arrest in human prostate cancer LNCaP cells. Carcinogenesis 2008, 29, 1049–1056. [Google Scholar] [CrossRef] [Green Version]
- Asadi-Samani, M.; Rafieian-Kopaei, M.; Lorigooini, Z.; Shirzad, H. A screening of growth inhibitory activity of Iranian medicinal plants on prostate cancer cell lines. BioMedicine 2018, 8. [Google Scholar] [CrossRef]
- Brower, V. Nutraceuticals: poised for a healthy slice of the healthcare market? Nature Biotechnol. 1998, 16, 728–731. [Google Scholar] [CrossRef]
- Higa, S.; Hirano, T.; Kotani, M.; Matsumoto, M.; Fujita, A.; Suemura, M.; Kawase, I.; Tanaka, T. Fisetin, a flavonol, inhibits T H 2-type cytokine production by activated human basophils. J. Allergy Clin. Immunol. 2003, 111, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Hewage, S.R.K.M.; Ryu, Y.S.; Oh, M.C.; Kwon, T.K.; Chae, S.; Hyun, J.W. Fisetin induces apoptosis and endoplasmic reticulum stress in human non-small cell lung cancer through inhibition of the MAPK signaling pathway. Tumor Biol. 2016, 37, 9615–9624. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. The J. Cell biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Brush, M.H.; Weiser, D.C.; Shenolikar, S.J.M. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1α to the endoplasmic reticulum and promotes dephosphorylation of the α subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 2003, 23, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Hafstad, A.D.; Beretta, M.; Zhang, M.; Molenaar, C.; Kopec, J.; Fotinou, D.; Murray, T.V.; Cobb, A.M.; Martin, D.J.T.E.j. Targeted redox inhibition of protein phosphatase 1 by Nox4 regulates eIF2α-mediated stress signaling. EMBO J. 2016, 35, 319–334. [Google Scholar] [CrossRef]
- Gyawali, B.; Prasad, V.J.N.R.C.O. Combining drugs and extending treatment—a PFS end point is not sufficient. Nat. Rev. Clin. Oncol. 2017, 14, 521. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, D.; Lee, Y. Metformin synergistically potentiates the antitumor effects of imatinib in colorectal cancer cells. Development Reprod. 2017, 21, 139. [Google Scholar]
- Su, C.H.; Kuo, C.L.; Lu, K.W.; Yu, F.S.; Ma, Y.S.; Yang, J.L.; Chu, Y.L.; Chueh, F.S.; Liu, K.C.; Chung, J.G. Fisetin-induced apoptosis of human oral cancer SCC-4 cells through reactive oxygen species production, endoplasmic reticulum stress, caspase-, and mitochondria-dependent signaling pathways. EnTox 2017, 32, 1725–1741. [Google Scholar] [CrossRef]
- Kamiya, T.; Hara, H.; Adachi, T. Effect of endoplasmic reticulum (ER) stress inducer thapsigargin on the expression of extracellular-superoxide dismutase in mouse 3T3-L1 adipocytes. J. Clin. Biochem. Nutr. 2013, 52, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-Y.; Kim, D.-S.; Auh, Q.-S.; Yi, J.-K.; Moon, S.U.; Kim, E.-C. Role of protein phosphatase 1 in angiogenesis and odontoblastic differentiation of human dental pulp cells. J. Endod. 2017, 43, 417–424. [Google Scholar] [CrossRef]
- Chalfant, C.E.; Kishikawa, K.; Mumby, M.C.; Kamibayashi, C.; Bielawska, A.; Hannun, Y.A. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A Activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem. 1999, 274, 20313–20317. [Google Scholar] [CrossRef]
- Luqmani, Y. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14, 35–48. [Google Scholar] [CrossRef]
- Veeresham, C. Natural products derived from plants as a source of drugs. In J. Adv. Pharm. Technol. Res.; 2012; Volume 3, pp. 200–201. [Google Scholar]
- Chou, R.-H.; Hsieh, S.-C.; Yu, Y.-L.; Huang, M.-H.; Huang, Y.-C.; Hsieh, Y.-H. Fisetin inhibits migration and invasion of human cervical cancer cells by down-regulating urokinase plasminogen activator expression through suppressing the p38 MAPK-dependent NF-κB signaling pathway. PloS One 2013, 8, e71983. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Lall, R.K.; Chamcheu, J.C.; Haidar, O.; Mukhtar, H. Involvement of ER stress and activation of apoptotic pathways in fisetin induced cytotoxicity in human melanoma. Arch. Biochem. Biophys. 2014, 563, 108–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Wang, Z.; Yang, J.; Zheng, W.; Chen, D.; Wu, G.; Sandford, R.; Tang, J.; Chen, X.-Z. Polycystin-1 inhibits eIF2α phosphorylation and cell apoptosis through a PKR-eIF2α pathway. Sci. Rep. 2017, 7, 11493. [Google Scholar] [CrossRef]
- Luo, W.; Xu, C.; Ayello, J.; Cruz, F.D.; Rosenblum, J.; Lessnick, S.; Cairo, M. Protein phosphatase 1 regulatory subunit 1A in ewing sarcoma tumorigenesis and metastasis. Oncogene 2018, 37, 798. [Google Scholar] [CrossRef] [PubMed]
- Grunicke, H.; Hofmann, J.; Utz, I. Role of protein kinases in antitumor drug resistance. Ann. Hematol. 1994, 69, S1–S6. [Google Scholar] [CrossRef]
- Fan, C.; Liu, Y.; Zhao, M.; Zhan, R.; Cui, W.; Jin, H.; Teng, Y.; Lv, P.; Zheng, L.; Huang, Y. Autophagy inhibits C2-ceramide-mediated cell death by decreasing the reactive oxygen species levels in SH-SY5Y cells. Neurosci. Lett. 2017, 651, 198–206. [Google Scholar] [CrossRef]
- Chou, H.-L.; Lin, Y.-H.; Liu, W.; Wu, C.-Y.; Li, R.-N.; Huang, H.-W.; Chou, C.-H.; Chiou, S.-J.; Chiu, C.-C. Combination Therapy of Chloroquine and C2-Ceramide Enhances Cytotoxicity in Lung Cancer H460 and H1299 Cells. Cancers 2019, 11, 370. [Google Scholar] [CrossRef]
- Jin, Z.; Zheng, L.; Xin, X.; Li, Y.; Hua, T.; Wu, T.; Wang, H. Upregulation of forkhead box O3 transcription is involved in C2-ceramide induced apoptosis and autophagy in ovarian cancer cells in vitro. Mol. Med. Rep. 2014, 10, 3099–3105. [Google Scholar] [CrossRef] [Green Version]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.-S.; Chang, Y.-C.; Kuo, W.-W.; Chen, M.-C.; Hsu, H.-H.; Tu, C.-C.; Yeh, Y.-L.; Viswanadha, V.P.; Liao, P.-H.; Huang, C.-Y. Inhibition of Protein Phosphatase 1 Stimulates Noncanonical ER Stress eIF2α Activation to Enhance Fisetin-induced Chemosensitivity in HDAC Inhibitor-resistant Hepatocellular Carcinoma Cells. Cancers 2019, 11, 918. https://doi.org/10.3390/cancers11070918
Liu Y-S, Chang Y-C, Kuo W-W, Chen M-C, Hsu H-H, Tu C-C, Yeh Y-L, Viswanadha VP, Liao P-H, Huang C-Y. Inhibition of Protein Phosphatase 1 Stimulates Noncanonical ER Stress eIF2α Activation to Enhance Fisetin-induced Chemosensitivity in HDAC Inhibitor-resistant Hepatocellular Carcinoma Cells. Cancers. 2019; 11(7):918. https://doi.org/10.3390/cancers11070918
Chicago/Turabian StyleLiu, Yi-Sheng, Yu-Chun Chang, Wei-Wen Kuo, Ming-Cheng Chen, Hsi-Hsien Hsu, Chuan-Chou Tu, Yu-Lan Yeh, Vijaya Padma Viswanadha, Po-Hsiang Liao, and Chih-Yang Huang. 2019. "Inhibition of Protein Phosphatase 1 Stimulates Noncanonical ER Stress eIF2α Activation to Enhance Fisetin-induced Chemosensitivity in HDAC Inhibitor-resistant Hepatocellular Carcinoma Cells" Cancers 11, no. 7: 918. https://doi.org/10.3390/cancers11070918
APA StyleLiu, Y. -S., Chang, Y. -C., Kuo, W. -W., Chen, M. -C., Hsu, H. -H., Tu, C. -C., Yeh, Y. -L., Viswanadha, V. P., Liao, P. -H., & Huang, C. -Y. (2019). Inhibition of Protein Phosphatase 1 Stimulates Noncanonical ER Stress eIF2α Activation to Enhance Fisetin-induced Chemosensitivity in HDAC Inhibitor-resistant Hepatocellular Carcinoma Cells. Cancers, 11(7), 918. https://doi.org/10.3390/cancers11070918