The Mode of Stem Cell Division Is Dependent on the Differential Interaction of β-Catenin with the Kat3 Coactivators CBP or p300

Abstract
:1. Introduction
2. Results
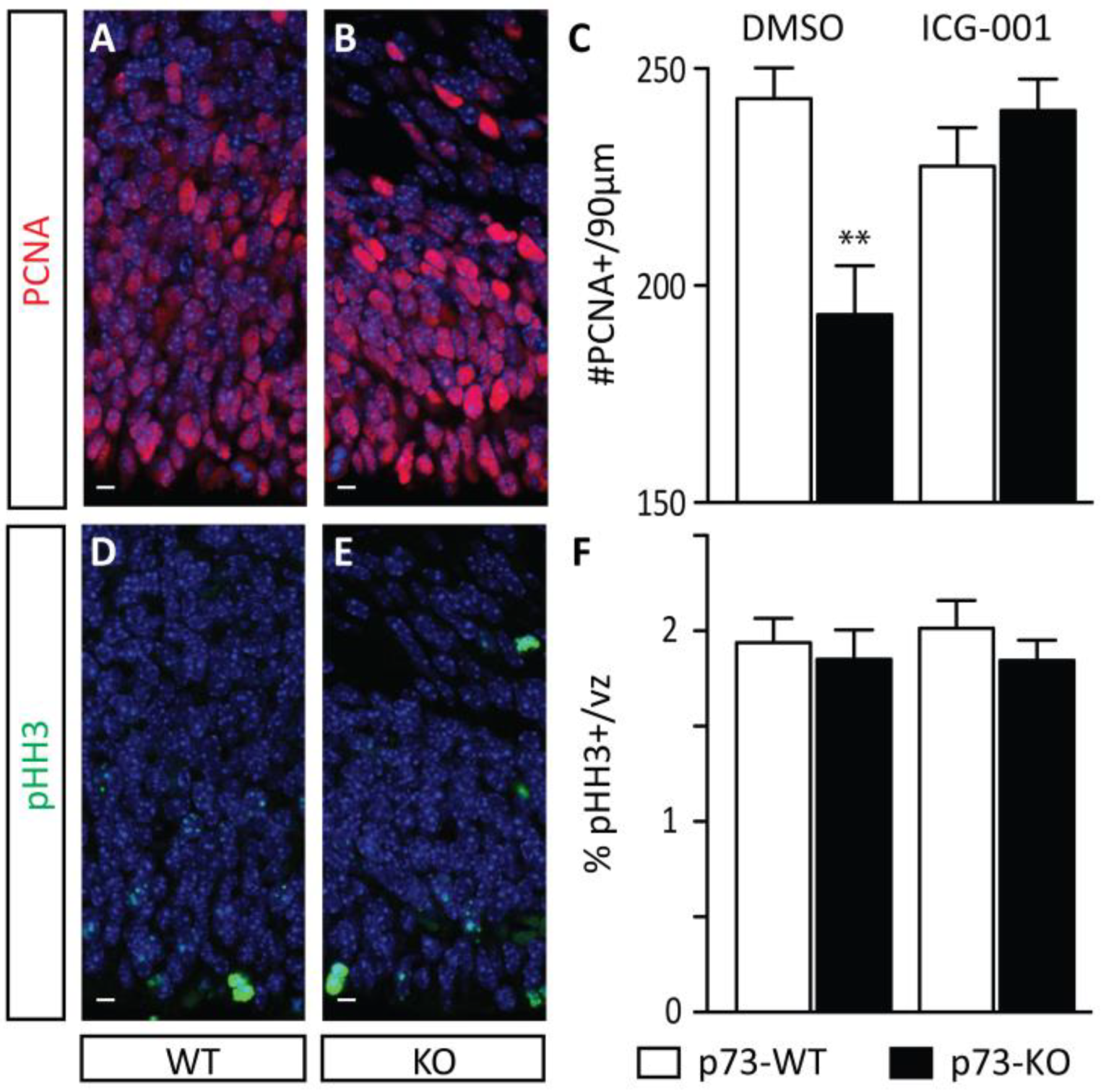
2.1. Increased Neuronal Production during the Early Stages of Corticogenesis Precedes the Depletion of the NSC Pool in p73KO Mice In Vivo
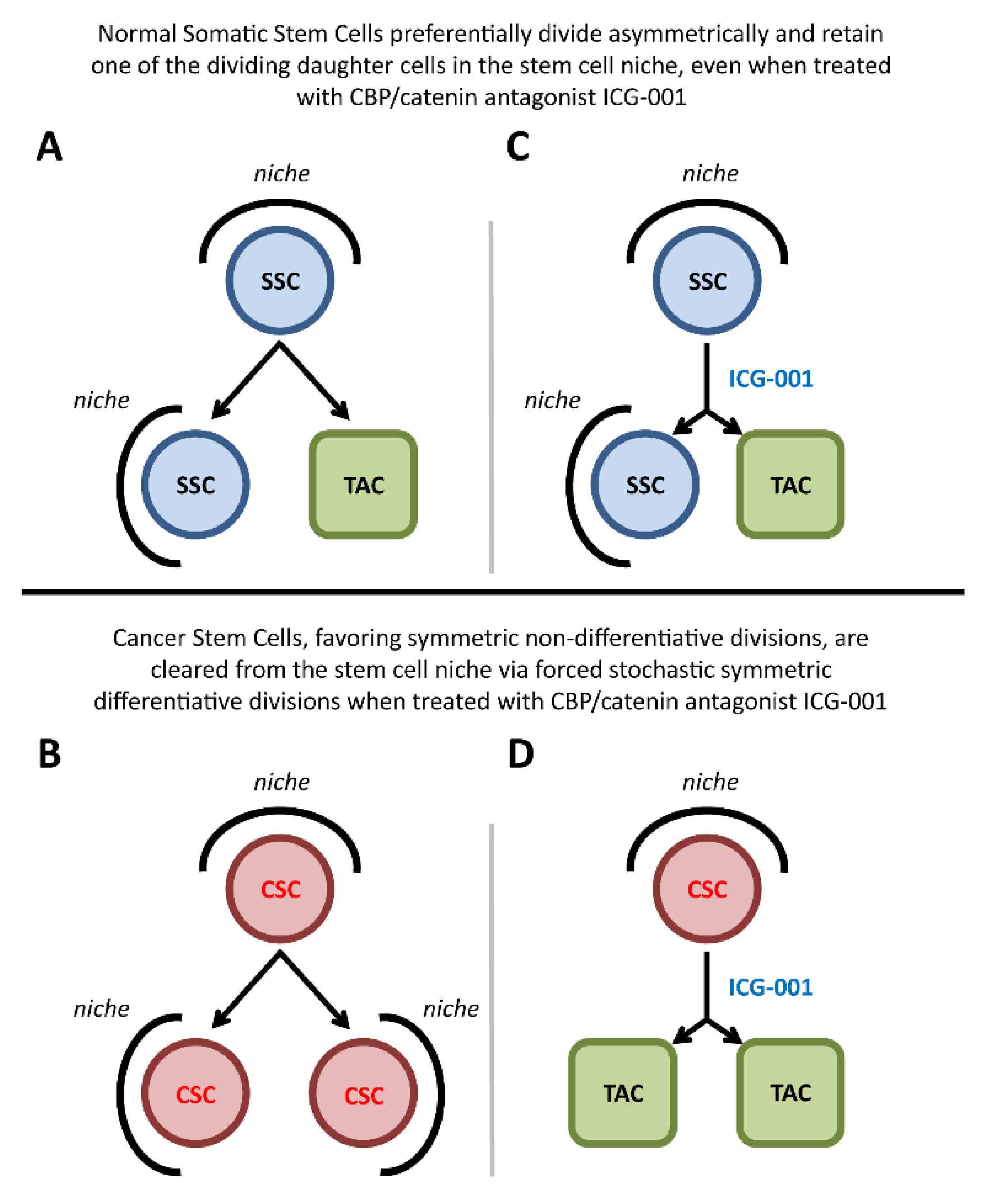
2.2. p73 Loss Induces A Decrease in the Proportion of Self-Renewing Asymmetric Divisions
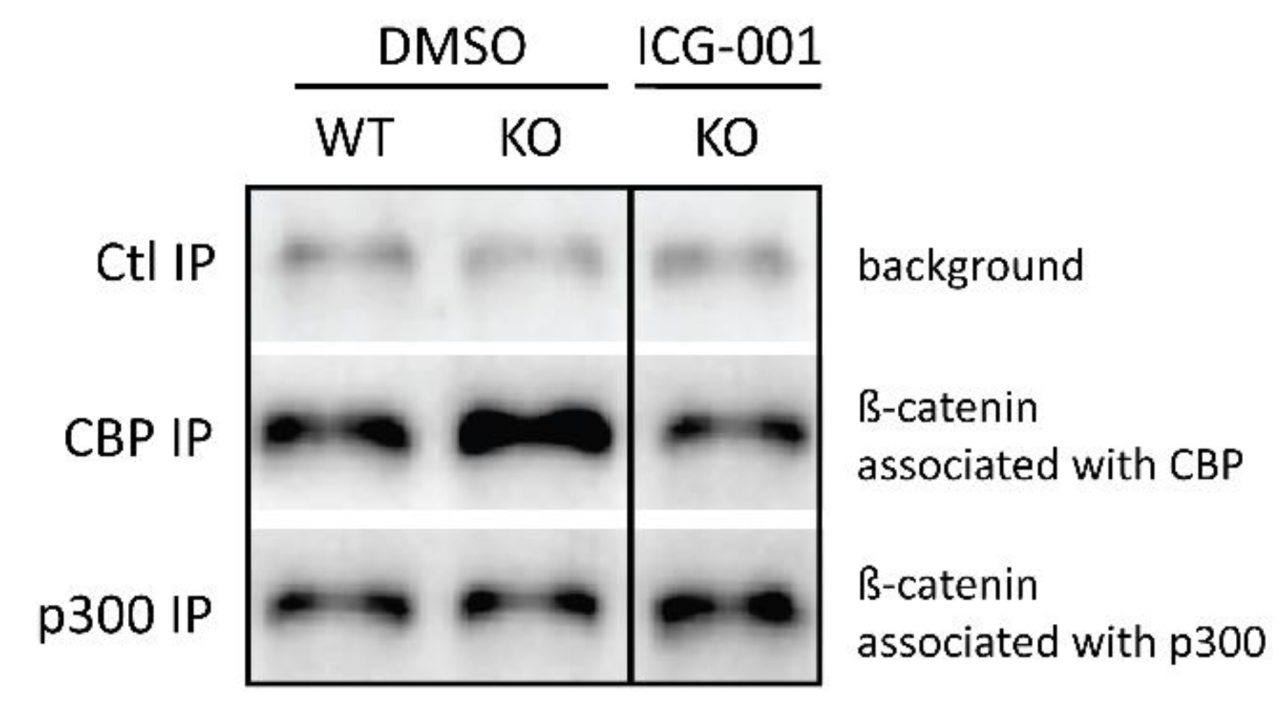
2.3. p73KO Neurogenic Phenotype Is Associated with An Increase in the CBP/β-Catenin Interaction
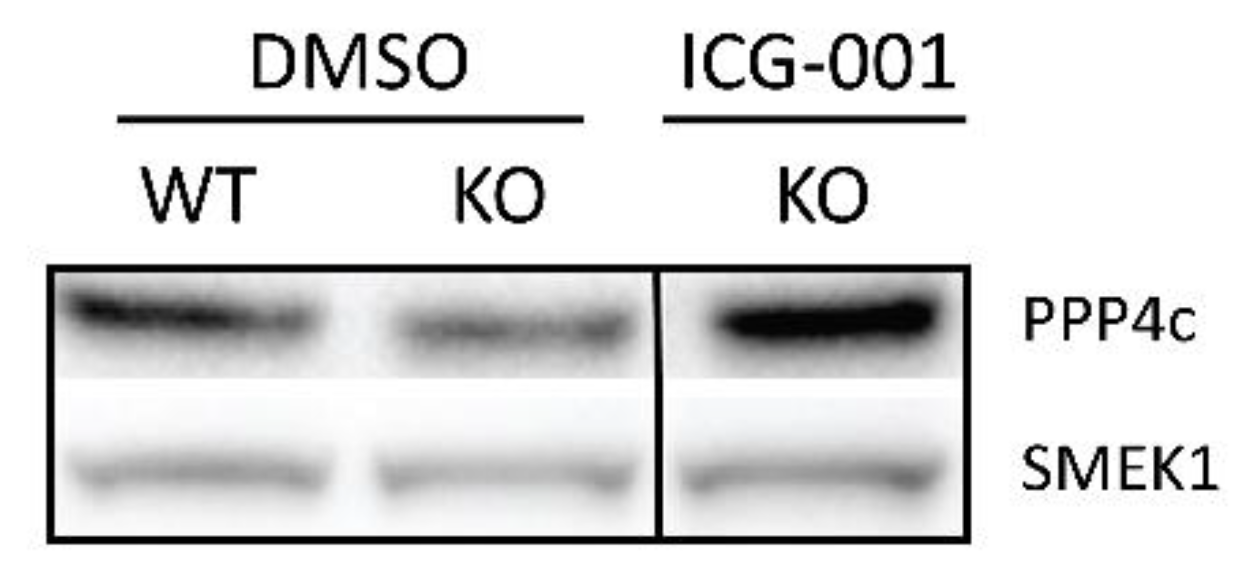
2.4. p73 Regulates PP4, A Key Factor in Mitotic Spindle Orientation
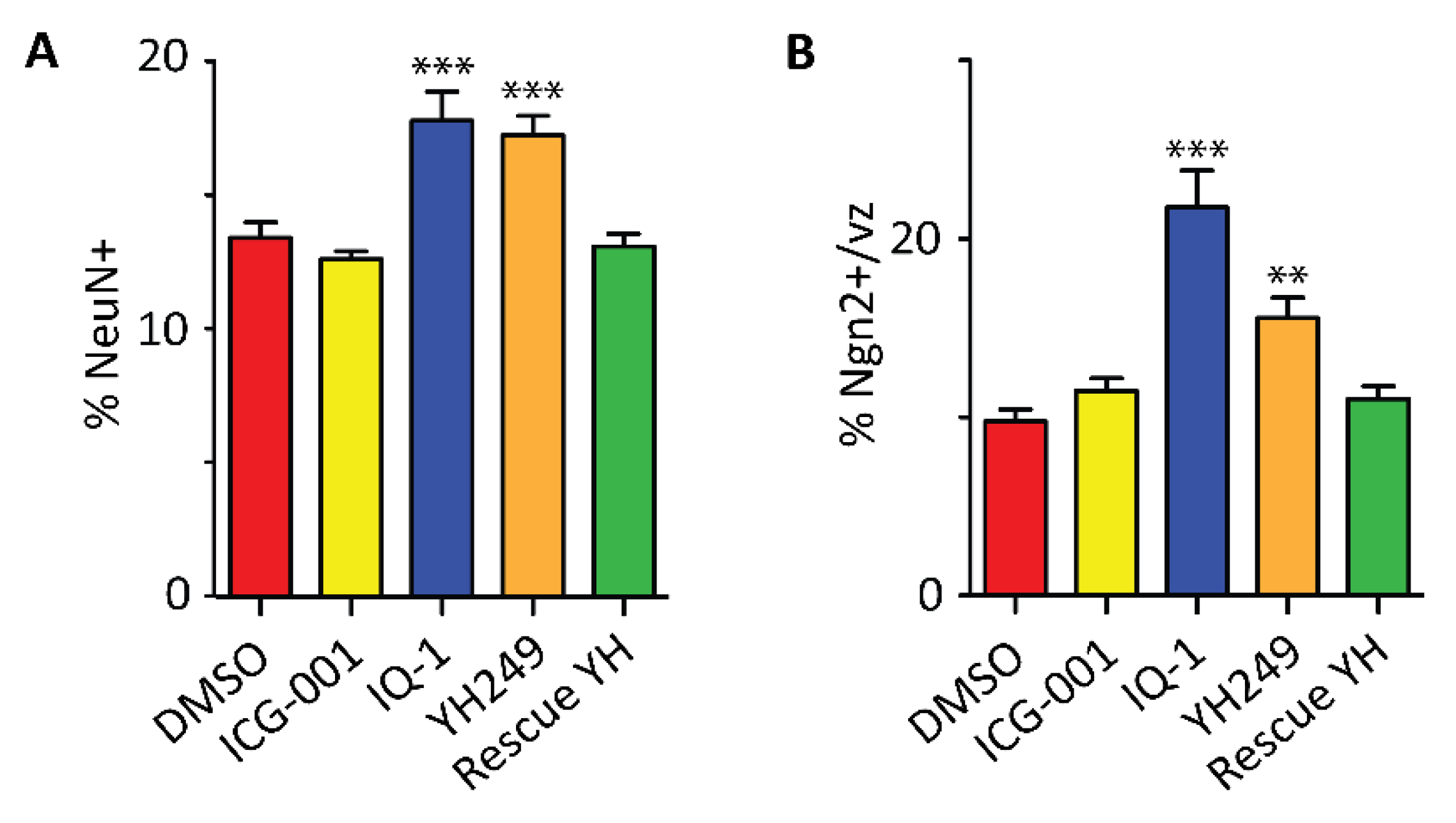
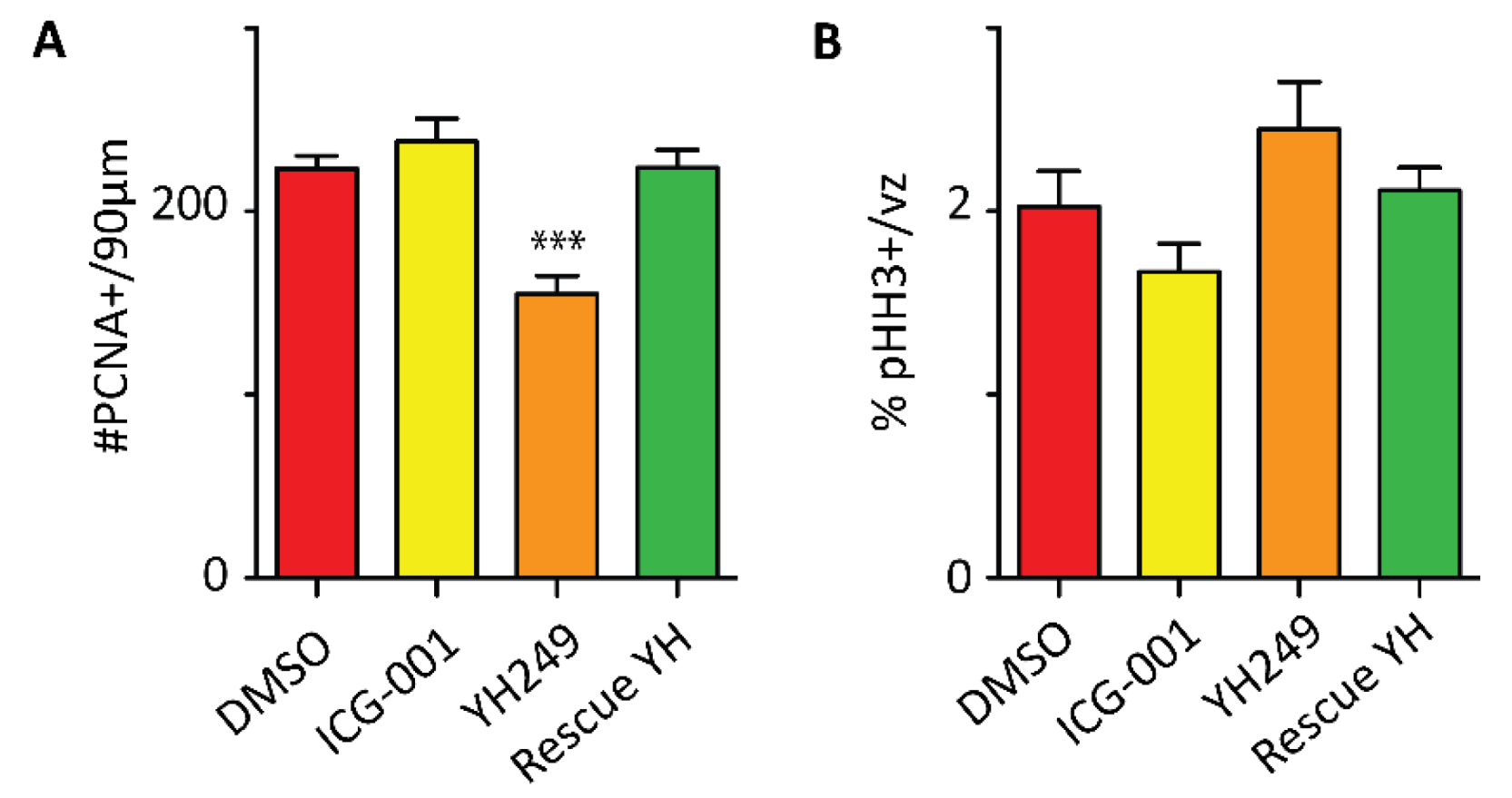
2.5. Specific Blockage of the p300/β-Catenin Interaction Recapitulates the p73KO Phenotype
3. Discussion
4. Materials and Methods
4.1. Mouse Lines and In Utero Treatment by Small Molecules
4.2. Immunofluorescence Staining
4.3. Western Blots
4.4. Par3 Quantification
4.5. Real-Time PCR (qPCR)
4.6. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kahn, M. Wnt Signaling in Stem Cells and Cancer Stem Cells: A Tale of Two Coactivators. Prog. Mol. Biol. Transl. Sci. 2018, 153, 209–244. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Kahn, M. Kat3 coactivators in somatic stem cells and cancer stem cells: Biological roles, evolution, and pharmacologic manipulation. Cell Biol. Toxicol. 2016, 32, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Wnt Signaling in Stem Cells and Tumor Stem Cells. Semin. Reprod. Med. 2015, 33, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Symmetric division versus asymmetric division: A tale of two coactivators. Future Med. Chem. 2011, 3, 1745–1763. [Google Scholar] [CrossRef] [PubMed]
- Manegold, P.; Lai, K.; Wu, Y.; Teo, J.L.; Lenz, H.J.; Genyk, Y.S.; Pandol, S.J.; Wu, K.; Lin, D.P.; Chen, Y.; et al. Differentiation Therapy Targeting the beta-Catenin/CBP Interaction in Pancreatic Cancer. Cancers 2018, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Masiello, D.; McMillian, M.; Nguyen, C.; Wu, Y.; Melendez, E.; Smbatyan, G.; Kida, A.; He, Y.; Teo, J.L.; et al. CBP/catenin antagonist safely eliminates drug-resistant leukemia-initiating cells. Oncogene 2016, 35, 3705–3717. [Google Scholar] [CrossRef] [PubMed]
- Gang, E.J.; Hsieh, Y.T.; Pham, J.; Zhao, Y.; Nguyen, C.; Huantes, S.; Park, E.; Naing, K.; Klemm, L.; Swaminathan, S.; et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 2014, 33, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- Wend, P.; Fang, L.; Zhu, Q.; Schipper, J.H.; Loddenkemper, C.; Kosel, F.; Brinkmann, V.; Eckert, K.; Hindersin, S.; Holland, J.D.; et al. Wnt/beta-catenin signalling induces MLL to create epigenetic changes in salivary gland tumours. EMBO J. 2013, 32, 1977–1989. [Google Scholar] [CrossRef]
- Sebio, A.; Kahn, M.; Lenz, H.J. The potential of targeting Wnt/beta-catenin in colon cancer. Expert Opin. Ther. Targets 2014, 18, 611–615. [Google Scholar] [CrossRef]
- Schade, B.; Lesurf, R.; Sanguin-Gendreau, V.; Bui, T.; Deblois, G.; O’Toole, S.A.; Millar, E.K.; Zardawi, S.J.; Lopez-Knowles, E.; Sutherland, R.L.; et al. Beta-Catenin signaling is a critical event in ErbB2-mediated mammary tumor progression. Cancer Res. 2013, 73, 4474–4487. [Google Scholar] [CrossRef]
- Park, E.; Gang, E.J.; Hsieh, Y.T.; Schaefer, P.; Chae, S.; Klemm, L.; Huantes, S.; Loh, M.; Conway, E.M.; Kang, E.S.; et al. Targeting survivin overcomes drug resistance in acute lymphoblastic leukemia. Blood 2011, 118, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, Y.; Hao, S.; Zhou, D.; Tan, R.J.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Multiple genes of the renin-angiotensin system are novel targets of Wnt/beta-catenin signaling. J. Am. Soc. Nephrol. JASN 2015, 26, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; He, W.; Li, Y.; Ding, H.; Hou, Y.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Targeted inhibition of beta-catenin/CBP signaling ameliorates renal interstitial fibrosis. J. Am. Soc. Nephrol. JASN 2011, 22, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Hwang, H.; Nguyen, C.; Kloner, R.A.; Kahn, M. The small molecule Wnt signaling modulator ICG-001 improves contractile function in chronically infarcted rat myocardium. PLoS ONE 2013, 8, e75010. [Google Scholar] [CrossRef] [PubMed]
- Schenke-Layland, K.; Nsair, A.; Van Handel, B.; Angelis, E.; Gluck, J.M.; Votteler, M.; Goldhaber, J.I.; Mikkola, H.K.; Kahn, M.; Maclellan, W.R. Recapitulation of the embryonic cardiovascular progenitor cell niche. Biomaterials 2011, 32, 2748–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, J.L.; Ma, H.; Nguyen, C.; Lam, C.; Kahn, M. Specific inhibition of CBP/beta-catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 12171–12176. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Gubbins, J.; Peng, Z.; Medina, V.; Fei, F.; Asahina, K.; Wang, J.; Kahn, M.; Rountree, C.B.; Stiles, B.L. Activation of hepatic stellate cell in Pten null liver injury model. Fibrogenesis Tissue Repair 2016, 9, 8. [Google Scholar] [CrossRef]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [Green Version]
- Lukaszewicz, A.I.; McMillan, M.K.; Kahn, M. Small molecules and stem cells. Potency and lineage commitment: The new quest for the fountain of youth. J. Med. Chem. 2010, 53, 3439–3453. [Google Scholar] [CrossRef]
- Noctor, S.C.; Martinez-Cerdeno, V.; Kriegstein, A.R. Contribution of intermediate progenitor cells to cortical histogenesis. Arch. Neurol. 2007, 64, 639–642. [Google Scholar] [CrossRef]
- Chenn, A.; McConnell, S.K. Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell 1995, 82, 631–641. [Google Scholar] [CrossRef]
- Kriegstein, A.; Noctor, S.; Martinez-Cerdeno, V. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat. Rev. Neurosci. 2006, 7, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Gotz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Pontious, A.; Kowalczyk, T.; Englund, C.; Hevner, R.F. Role of intermediate progenitor cells in cerebral cortex development. Dev. Neurosci. 2008, 30, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Verne S, C.; Takahashi, T. Proliferative events in the cerebral ventricular zone. Brain Dev. 1995, 17, 159–163. [Google Scholar]
- Rakic, P. A small step for the cell, a giant leap for mankind: A hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995, 18, 383–388. [Google Scholar] [CrossRef]
- Chenn, A.; Walsh, C.A. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 2002, 297, 365–369. [Google Scholar] [CrossRef]
- Gilmore, E.C.; Walsh, C.A. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip. Rev. Dev. Biol 2013, 2, 461–478. [Google Scholar] [CrossRef]
- Yadlapalli, S.; Yamashita, Y.M. Chromosome-specific nonrandom sister chromatid segregation during stem-cell division. Nature 2013, 498, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Chia, W. Neurogenesis and asymmetric cell division. Curr. Opinion Neurobiol. 2008, 18, 4–11. [Google Scholar] [CrossRef]
- Fish, J.L.; Kosodo, Y.; Enard, W.; Paabo, S.; Huttner, W.B. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 10438–10443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanada, K.; Tsai, L.H. G protein betagamma subunits and AGS3 control spindle orientation and asymmetric cell fate of cerebral cortical progenitors. Cell 2005, 122, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Walsh, C.A. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 2004, 44, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Li, M.; Lin, Q.; Qi, X.; Su, B. Functional divergence of the brain-size regulating gene MCPH1 during primate evolution and the origin of humans. BMC Biol. 2013, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Killick, R.; Niklison-Chirou, M.; Tomasini, R.; Bano, D.; Rufini, A.; Grespi, F.; Velletri, T.; Tucci, P.; Sayan, B.S.; Conforti, F.; et al. p73: A multifunctional protein in neurobiology. Molecular Neurobiol. 2011, 43, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Acosta, N.C.; Cabrera-Socorro, A.; Morlans, M.P.; Delgado, F.J.; Suarez-Sola, M.L.; Sottocornola, R.; Lu, X.; Gonzalez-Gomez, M.; Meyer, G. Dynamic expression of the p53 family members p63 and p73 in the mouse and human telencephalon during development and in adulthood. Brain Res. 2011, 1372, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Tucci, P.; Chen, H.; Knight, R.A.; Bano, D.; Nicotera, P.; McKeon, F.; Melino, G. P73 regulates maintenance of neural stem cell. Biochem. Biophys. Res. Commun. 2010, 403, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cano, L.; Herreros-Villanueva, M.; Fernandez-Alonso, R.; Ayuso-Sacido, A.; Meyer, G.; Garcia-Verdugo, J.M.; Silva, A.; Marques, M.M.; Marin, M.C. P73 deficiency results in impaired self renewal and premature neuronal differentiation of mouse neural progenitors independently of p53. Cell Death Dis. 2010, 1, e109. [Google Scholar] [CrossRef] [PubMed]
- Talos, F.; Abraham, A.; Vaseva, A.V.; Holembowski, L.; Tsirka, S.E.; Scheel, A.; Bode, D.; Dobbelstein, M.; Bruck, W.; Moll, U.M. P73 is an essential regulator of neural stem cell maintenance in embryonal and adult CNS neurogenesis. Cell Death Differ. 2010, 17, 1816–1829. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, M.; Cancino, G.I.; Dugani, C.B.; Weaver, I.C.; Gauthier-Fisher, A.; Paquin, A.; Mak, T.W.; Wojtowicz, M.J.; Miller, F.D.; Kaplan, D.R. TAp73 acts via the bHLH Hey2 to promote long-term maintenance of neural precursors. Curr. Biol. CB 2010, 20, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Talos, F.; Moll, U.M. P73 is dispensable for commitment to neural stem cell fate, but is essential for neural stem cell maintenance and for blocking premature differentiation. Cell Death Differ. 2013, 20, 368. [Google Scholar] [CrossRef] [PubMed]
- Papaspyropoulos, A.; Bradley, L.; Thapa, A.; Leung, C.Y.; Toskas, K.; Koennig, D.; Pefani, D.E.; Raso, C.; Grou, C.; Hamilton, G.; et al. RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat. Commun. 2018, 9, 424. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Hijikata, M.; Takagi, S.; Takada, R.; Takada, S.; Chiba, T.; Shimotohno, K. P73beta, a variant of p73, enhances Wnt/beta-catenin signaling in Saos-2 cells. Biochem. Biophys. Res Commun. 2001, 283, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.S.; Gilliland, D.G.; Morrison, S.J. MTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 2010, 7, 593–605. [Google Scholar] [CrossRef]
- Urban, N.; Guillemot, F. Neurogenesis in the embryonic and adult brain: Same regulators, different roles. Front. Cell. Neurosci. 2014, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Bultje, R.S.; Castaneda-Castellanos, D.R.; Jan, L.Y.; Jan, Y.N.; Kriegstein, A.R.; Shi, S.H. Mammalian Par3 regulates progenitor cell asymmetric division via notch signaling in the developing neocortex. Neuron 2009, 63, 189–202. [Google Scholar] [CrossRef]
- Haydar, T.F.; Ang, E.; Rakic, P. Mitotic spindle rotation and mode of cell division in the developing telencephalon. Proc. Natl. Acad. Sci. USA 2003, 100, 2890–2895. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, K.; Wuttke, A.; Quadrato, G.; Chumakov, P.M.; Wizenmann, A.; Di Giovanni, S. The tumor suppressor p53 fine-tunes reactive oxygen species levels and neurogenesis via PI3 kinase signaling. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 14318–14330. [Google Scholar] [CrossRef]
- Medina-Bolivar, C.; Gonzalez-Arnay, E.; Talos, F.; Gonzalez-Gomez, M.; Moll, U.M.; Meyer, G. Cortical hypoplasia and ventriculomegaly of p73-deficient mice: Developmental and adult analysis. J. Comp. Neurol. 2014, 522, 2663–2679. [Google Scholar] [CrossRef]
- Pozniak, C.D.; Radinovic, S.; Yang, A.; McKeon, F.; Kaplan, D.R.; Miller, F.D. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 2000, 289, 304–306. [Google Scholar] [CrossRef]
- Miyabayashi, T.; Teo, J.L.; Yamamoto, M.; McMillan, M.; Nguyen, C.; Kahn, M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 2007, 104, 5668–5673. [Google Scholar] [CrossRef]
- Hasegawa, K.; Yasuda, S.Y.; Teo, J.L.; Nguyen, C.; McMillan, M.; Hsieh, C.L.; Suemori, H.; Nakatsuji, N.; Yamamoto, M.; Miyabayashi, T.; et al. Wnt signaling orchestration with a small molecule DYRK inhibitor provides long-term xeno-free human pluripotent cell expansion. Stem Cells Trans. Med. 2012, 1, 18–28. [Google Scholar] [CrossRef]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.L.; Oh, S.W.; Kim, H.Y.; et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef]
- Ma, H.; Nguyen, C.; Lee, K.S.; Kahn, M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene 2005, 24, 3619–3631. [Google Scholar] [CrossRef]
- Das, R.M.; Storey, K.G. Mitotic spindle orientation can direct cell fate and bias Notch activity in chick neural tube. EMBO Rep. 2012, 13, 448–454. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Spindle orientation in mammalian cerebral cortical development. Curr. Opin. Neurobiol. 2012, 22, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Morin, X.; Bellaiche, Y. Mitotic spindle orientation in asymmetric and symmetric cell divisions during animal development. Dev. Cell 2011, 21, 102–119. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Marrocco, C.; Rinalducci, S.; Peschiaroli, A.; Timperio, A.M.; Bongiorno-Borbone, L.; Finazzi Agro, A.; Melino, G.; Zolla, L. Analysis of TAp73-dependent signaling via omics technologies. J. Proteome Res. 2013, 12, 4207–4220. [Google Scholar] [CrossRef]
- Xie, Y.; Juschke, C.; Esk, C.; Hirotsune, S.; Knoblich, J.A. The phosphatase PP4c controls spindle orientation to maintain proliferative symmetric divisions in the developing neocortex. Neuron 2013, 79, 254–265. [Google Scholar] [CrossRef]
- Lyu, J.; Kim, H.R.; Yamamoto, V.; Choi, S.H.; Wei, Z.; Joo, C.K.; Lu, W. Protein phosphatase 4 and Smek complex negatively regulate Par3 and promote neuronal differentiation of neural stem/progenitor cells. Cell Rep. 2013, 5, 593–600. [Google Scholar] [CrossRef]
- Pawlisz, A.S.; Mutch, C.; Wynshaw-Boris, A.; Chenn, A.; Walsh, C.A.; Feng, Y. Lis1-Nde1-dependent neuronal fate control determines cerebral cortical size and lamination. Hum. Mol. Genet. 2008, 17, 2441–2455. [Google Scholar] [CrossRef]
- Sousa-Nunes, R.; Chia, W.; Somers, W.G. Protein phosphatase 4 mediates localization of the Miranda complex during Drosophila neuroblast asymmetric divisions. Genes Dev. 2009, 23, 359–372. [Google Scholar] [CrossRef]
- Lyu, J.; Jho, E.H.; Lu, W. Smek promotes histone deacetylation to suppress transcription of Wnt target gene brachyury in pluripotent embryonic stem cells. Cell Res. 2011, 21, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Acebron, S.P.; Niehrs, C. β-Catenin-Independent Roles of Wnt/LRP6 Signaling. Trends Cell Biol. 2016, 26, 956–967. [Google Scholar] [CrossRef]
- Higuchi, Y.; Nguyen, C.; Yasuda, S.Y.; McMillan, M.; Hasegawa, K.; Kahn, M. Specific Direct Small Molecule p300/beta-Catenin Antagonists Maintain Stem Cell Potency. Curr. Mol. Pharmacol. 2016, 9, 272–279. [Google Scholar] [CrossRef]
- Rieger, M.E.; Zhou, B.; Solomon, N.; Sunohara, M.; Li, C.; Nguyen, C.; Liu, Y.; Pan, J.H.; Minoo, P.; Crandall, E.D.; et al. p300/beta-Catenin Interactions Regulate Adult Progenitor Cell Differentiation Downstream of WNT5a/Protein Kinase C (PKC). J. Biol. Chem. 2016, 291, 6569–6582. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, K.; Nguyen, C.; Smbatyan, G.; Melendez, E.; Higuchi, Y.; Chen, Y.; Kahn, M. Small molecule p300/catenin antagonist enhances hematopoietic recovery after radiation. PLoS ONE 2017, 12, e0177245. [Google Scholar] [CrossRef]
- Haubensak, W.; Attardo, A.; Denk, W.; Huttner, W.B. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: A major site of neurogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 3196–3201. [Google Scholar] [CrossRef] [Green Version]
- Attardo, A.; Fabel, K.; Krebs, J.; Haubensak, W.; Huttner, W.B.; Kempermann, G. Tis21 expression marks not only populations of neurogenic precursor cells but also new postmitotic neurons in adult hippocampal neurogenesis. Cereb. Cortex 2010, 20, 304–314. [Google Scholar] [CrossRef]
- Falk, S.; Bugeon, S.; Ninkovic, J.; Pilz, G.A.; Postiglione, M.P.; Cremer, H.; Knoblich, J.A.; Gotz, M. Time-Specific Effects of Spindle Positioning on Embryonic Progenitor Pool Composition and Adult Neural Stem Cell Seeding. Neuron 2017, 93, 777–791. [Google Scholar] [CrossRef]
- Huttner, W.B.; Kosodo, Y. Symmetric versus asymmetric cell division during neurogenesis in the developing vertebrate central nervous system. Curr. Opin. Cell Biol. 2005, 17, 648–657. [Google Scholar] [CrossRef]
- Takahashi, T.; Nowakowski, R.S.; Caviness, V.S. The leaving or Q fraction of the murine cerebral proliferative epithelium: A general model of neocortical neuronogenesis. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 6183–6196. [Google Scholar] [CrossRef]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef]
- Zinin, N.; Adameyko, I.; Wilhelm, M.; Fritz, N.; Uhlen, P.; Ernfors, P.; Henriksson, M.A. MYC proteins promote neuronal differentiation by controlling the mode of progenitor cell division. EMBO Rep. 2014, 15, 383–391. [Google Scholar] [CrossRef]
- Israsena, N.; Hu, M.; Fu, W.; Kan, L.; Kessler, J.A. The presence of FGF2 signaling determines whether beta-catenin exerts effects on proliferation or neuronal differentiation of neural stem cells. Dev. Biol. 2004, 268, 220–231. [Google Scholar] [CrossRef]
- Sanchez-Taltavull, D. Optimal architecture of differentiation cascades with asymmetric and symmetric stem cell division. J. Theor. Biol. 2016, 407, 106–117. [Google Scholar] [CrossRef]
- McCrea, P.D.; Gottardi, C.J. Beyond beta-catenin: Prospects for a larger catenin network in the nucleus. Nat. Rev. Mol. Cell Biol. 2016, 17, 55–64. [Google Scholar] [CrossRef]
- Gottardi, C.J.; Konigshoff, M. Considerations for targeting beta-catenin signaling in fibrosis. Am. J. Respir. Crit. Care Med. 2013, 187, 566–568. [Google Scholar] [CrossRef]
- Magee, J.A.; Ikenoue, T.; Nakada, D.; Lee, J.Y.; Guan, K.L.; Morrison, S.J. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 2012, 11, 415–428. [Google Scholar] [CrossRef]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef]
- Reiner, O.; Sapir, T. LIS1 functions in normal development and disease. Curr. Opin. Neurobiol. 2013, 23, 951–956. [Google Scholar] [CrossRef]
- Zimdahl, B.; Ito, T.; Blevins, A.; Bajaj, J.; Konuma, T.; Weeks, J.; Koechlein, C.S.; Kwon, H.Y.; Arami, O.; Rizzieri, D.; et al. Lis1 regulates asymmetric division in hematopoietic stem cells and in leukemia. Nat. Genet. 2014, 46, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009, 138, 1083–1095. [Google Scholar] [CrossRef]
- Hwang, W.L.; Jiang, J.K.; Yang, S.H.; Huang, T.S.; Lan, H.Y.; Teng, H.W.; Yang, C.Y.; Tsai, Y.P.; Lin, C.H.; Wang, H.W.; et al. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat. Cell Biol. 2014, 16, 268–280. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | # Experiments (# Embryos) | # Mitoses | % Asm | Statistic |
|---|---|---|---|---|
| DMSO-WT | 3(7) | 63 | 41.3 | (a) |
| DMSO-KO | 3(6) | 66 | 16.7 | * |
| ICG-001-WT | 3(10) | 74 | 40.5 | (a) |
| ICG-001-KO | 3(10) | 72 | 45.8 | ns |
| Condition | # Experiments (# embryos) | # Mitoses | %Asm | Statistic |
|---|---|---|---|---|
| DMSO-WT | 6(17) | 153 | 46.4 | (a) |
| ICG-001 (3x)-WT | 3(11) | 87 | 47.1 | ns |
| YH249-WT | 4(11) | 87 | 6.9 | ** |
| YH249 Rescue-WT | 3(7) | 52 | 36.5 | ns |
| IQ-1-WT | 4(11) | 90 | 22.2 | * |
| DMSO-Tis21-GFP | 4(7) | 32 | 65.6 | (b) |
| YH249-Tis21-GFP | 3(8) | 29 | 56.7 | ns |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lukaszewicz, A.I.; Nguyen, C.; Melendez, E.; Lin, D.P.; Teo, J.-L.; Lai, K.K.Y.; Huttner, W.B.; Shi, S.-H.; Kahn, M. The Mode of Stem Cell Division Is Dependent on the Differential Interaction of β-Catenin with the Kat3 Coactivators CBP or p300. Cancers 2019, 11, 962. https://doi.org/10.3390/cancers11070962
Lukaszewicz AI, Nguyen C, Melendez E, Lin DP, Teo J-L, Lai KKY, Huttner WB, Shi S-H, Kahn M. The Mode of Stem Cell Division Is Dependent on the Differential Interaction of β-Catenin with the Kat3 Coactivators CBP or p300. Cancers. 2019; 11(7):962. https://doi.org/10.3390/cancers11070962
Chicago/Turabian StyleLukaszewicz, Agnes I., Cu Nguyen, Elizabeth Melendez, David P. Lin, Jia-Ling Teo, Keane K. Y. Lai, Wieland B. Huttner, Song-Hai Shi, and Michael Kahn. 2019. "The Mode of Stem Cell Division Is Dependent on the Differential Interaction of β-Catenin with the Kat3 Coactivators CBP or p300" Cancers 11, no. 7: 962. https://doi.org/10.3390/cancers11070962
APA StyleLukaszewicz, A. I., Nguyen, C., Melendez, E., Lin, D. P., Teo, J. -L., Lai, K. K. Y., Huttner, W. B., Shi, S. -H., & Kahn, M. (2019). The Mode of Stem Cell Division Is Dependent on the Differential Interaction of β-Catenin with the Kat3 Coactivators CBP or p300. Cancers, 11(7), 962. https://doi.org/10.3390/cancers11070962