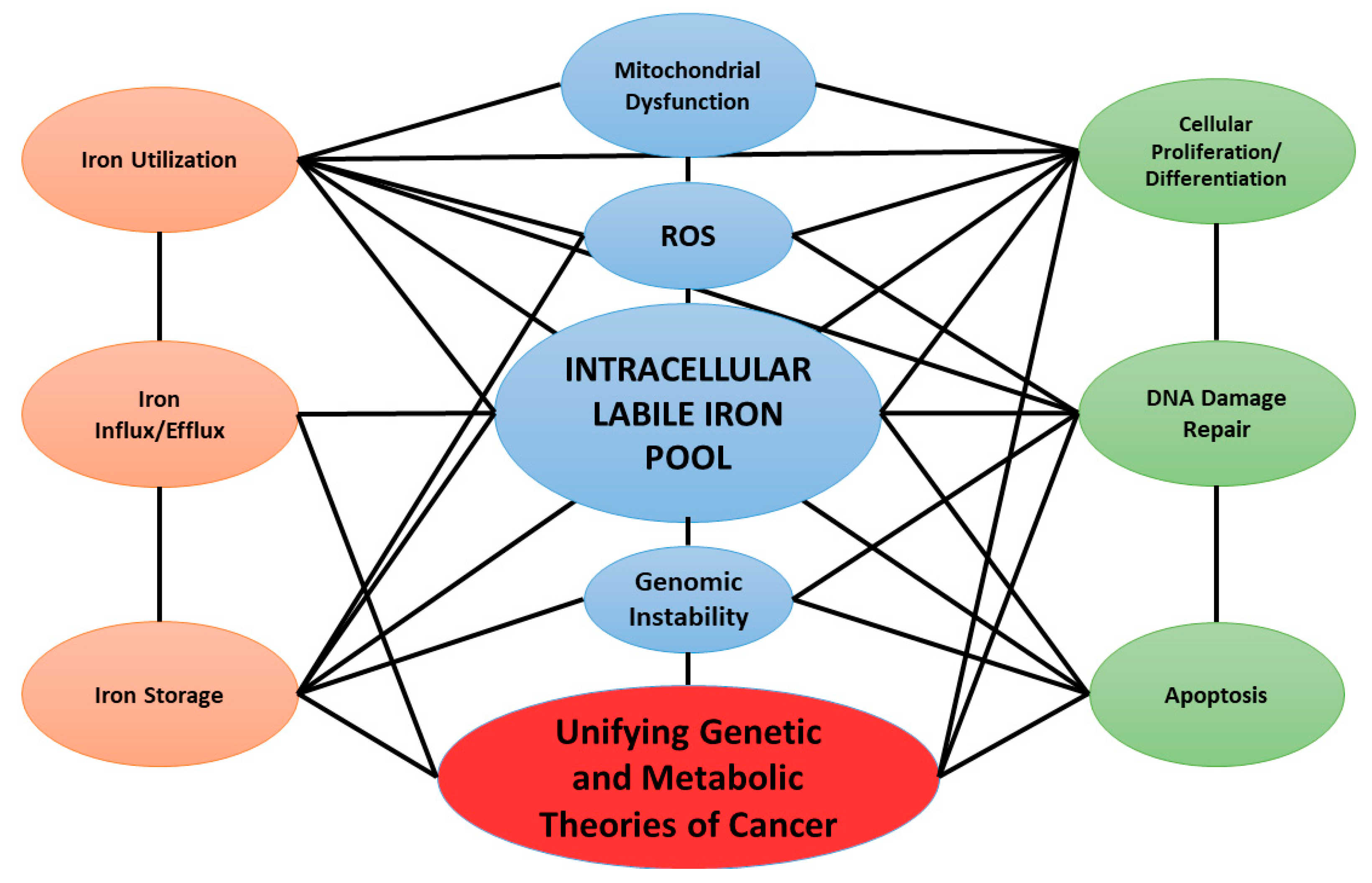
Linking Cancer Metabolic Dysfunction and Genetic Instability through the Lens of Iron Metabolism



Abstract
:1. Introduction
2. Overview of Intracellular Iron Metabolism
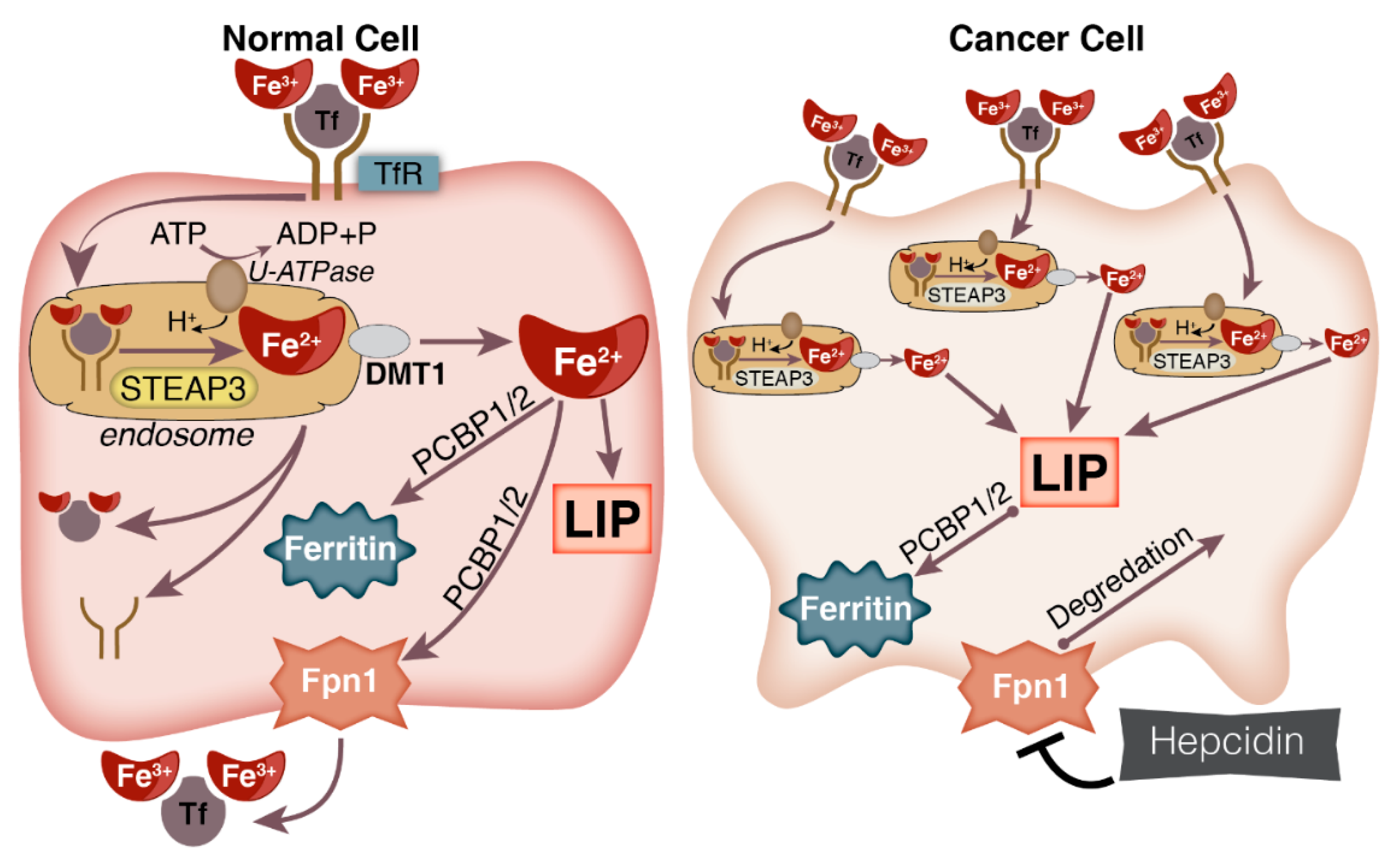
2.1. Iron Uptake, Storage, and Homeostasis
2.2. Mitochondrial Iron Metabolism
2.3. Heme Synthesis
2.4. Iron Sulfur Cluster Biogenesis
2.5. Mitochondrial Iron Storage
3. Iron and Cancer
3.1. Iron and Traditional Hallmarks of Cancer
3.2. Iron Influx/Efflux and Cancer
3.3. Mitochondrial Iron Metabolism and Cancer
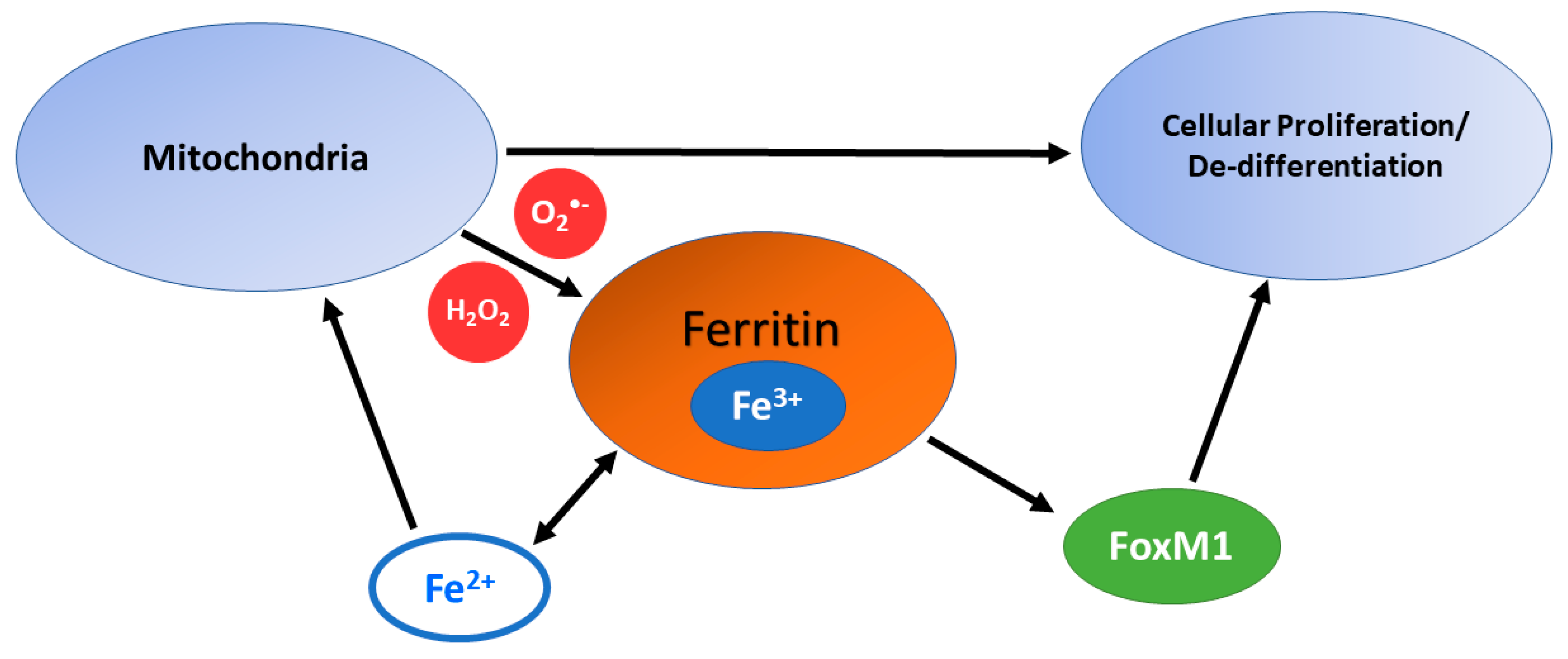
3.4. Mitochondrial Dysfunction, ROS, and Iron Storage in Carcinogenesis
4. Targeting Iron in Cancer
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Troadec, M.-B.; Courselaud, B.; Détivaud, L.; Haziza-Pigeon, C.; Leroyer, P.; Brissot, P.; Loréal, O. Iron Overload Promotes Cyclin D1 Expression and Alters Cell Cycle in Mouse Hepatocytes. J. Hepatol. 2006, 44, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Hinchliffe, P.; Sazanov, L.A. Organization of Iron-Sulfur Clusters in Respiratory Complex I. Science 2005, 309, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. P21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, M. Labile Iron Pool: The Main Determinant of Cellular Response to Oxidative Stress. Oxid. DNA Damage Repair Base Excision Repair 2003, 531, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Breuer, W.; Shvartsman, M.; Cabantchik, Z.I. Intracellular Labile Iron. Int. J. Biochem. Cell Biol. 2008, 40, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.Y.; Buettner, G.R. Iron and Dioxygen Chemistry Is an Important Route to Initiation of Biological Free Radical Oxidations: An Electron Paramagnetic Resonance Spin Trapping Study. Free Radic. Biol. Med. 1999, 26, 1447–1456. [Google Scholar] [CrossRef]
- Wardman, P.; Candeias, L.P. Fenton Chemistry: An Introduction. Radiat. Res. 1996, 145, 523–531. [Google Scholar] [CrossRef]
- Dev, S.; Babitt, J.L. Overview of Iron Metabolism in Health and Disease. Hemodial. Int. Int. Symp. Home Hemodial. 2017, 21, S6–S20. [Google Scholar] [CrossRef]
- Li, K.; Reichmann, H. Role of Iron in Neurodegenerative Diseases. J. Neural Transm. 2016, 123, 389–399. [Google Scholar] [CrossRef]
- Camaschella, C. Iron-Deficiency Anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef]
- Miller, D.M.; Buettner, G.R.; Aust, S.D. Transition Metals as Catalysts of “Autoxidation” Reactions. Free Radic. Biol. Med. 1990, 8, 95–108. [Google Scholar] [CrossRef]
- Linberg, R.; Conover, C.; Shum, K.; Shorr, R. Hemoglobin Based Oxygen Carriers: How Much Methemoglobin Is Too Much? Artif Cells Blood Substit. Immobil Biotechnol. 1998, 26, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Beinert, H. Iron-Sulfur Proteins: Ancient Structures, Still Full of Surprises. JBIC J. Biol. Inorg. Chem. 2000, 5, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Keyer, K.; Imlay, J.A. Superoxide Accelerates DNA Damage by Elevating Free-Iron Levels. Proc. Natl. Acad. Sci. USA 1996, 93, 13635–13640. [Google Scholar] [CrossRef] [PubMed]
- Piñero, D.J.; Connor, J.R. Iron in the Brain: An Important Contributor in Normal and Diseased States. Neuroscientist 2000, 6, 435–453. [Google Scholar] [CrossRef]
- Silva, B.; Faustino, P. An Overview of Molecular Basis of Iron Metabolism Regulation and the Associated Pathologies. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2015, 1852, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, L.; Ding, J.; Chen, Y. Iron Metabolism in Cancer. Int. J. Mol. Sci. 2018, 20, 95. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Ironing out Cancer. Cancer Res. 2011, 71, 1511–1514. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and Cancer: More Ore to Be Mined. Nat. Rev. Cancer 2013, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Chekhun, S.; Lukyanova, N.; Shvets, Y.; Burlaka, A.; Buchynska, L. Significance of Ferritin Expression in Formation of Malignant Phenotype of Human Breast Cancer Cells. Exp. Oncol. 2014, 36, 179–183. [Google Scholar]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2•− and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, R.J.; David Andros, N.; Watt, R.K. The Ferroxidase Center Is Essential for Ferritin Iron Loading in the Presence of Phosphate and Minimizes Side Reactions That Form Fe(III)-Phosphate Colloids. BioMetals 2012, 25, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Bakker, G.; Boyer, R. Iron Incorporation into Apoferritin. The Role of Apoferritin as a Ferroxidase. J. Biol. Chem. 1986, 261, 13182–13185. [Google Scholar] [PubMed]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A Cytosolic Iron Chaperone That Delivers Iron to Ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, A.G.; Nandal, A.; Park, J.H.; Smith, P.M.; Yabe, T.; Ryu, M.-S.; Ghosh, M.C.; Lee, J.; Rouault, T.A.; Park, M.H.; et al. Iron Chaperones PCBP1 and PCBP2 Mediate the Metallation of the Dinuclear Iron Enzyme Deoxyhypusine Hydroxylase. Proc. Natl. Acad. Sci. USA 2014, 111, 8031. [Google Scholar] [CrossRef]
- Yanatori, I.R.; Richardson, D.; Imada, K.; Kishi, F. Iron Export through the Transporter Ferroportin 1 Is Modulated by the Iron Chaperone PCBP2. J. Biol. Chem. 2016, 291. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, L.; Xiao, L.; Liu, R.; Li, Z.; Wang, Y. The RNA-Binding Protein PCBP1 Functions as a Tumor Suppressor in Prostate Cancer by Inhibiting Mitogen Activated Protein Kinase 1. Cell. Physiol. Biochem. 2018, 48, 1747–1754. [Google Scholar] [CrossRef]
- Cairo, G.; Pietrangelo, A. Iron Regulatory Proteins in Pathobiology. Biochem. J. 2000, 352 Pt 2, 241–250. [Google Scholar] [CrossRef]
- Takahashi-Makise, N.; Ward, D.M.; Kaplan, J. On the Mechanism of Iron Sensing by IRP2: New Players, New Paradigms. Nat. Chem. Biol. 2009, 5, 874–875. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of Mitochondrial Iron Import through Differential Turnover of Mitoferrin 1 and Mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troadec, M.-B.; Warner, D.; Wallace, J.; Thomas, K.; Spangrude, G.J.; Phillips, J.; Khalimonchuk, O.; Paw, B.H.; Ward, D.M.; Kaplan, J. Targeted Deletion of the Mouse Mitoferrin1 Gene: From Anemia to Protoporphyria. Blood 2011, 117, 5494–5502. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Paradkar, P.N.; Li, L.; Pierce, E.L.; Langer, N.B.; Takahashi-Makise, N.; Hyde, B.B.; Shirihai, O.S.; Ward, D.M.; Kaplan, J.; et al. Abcb10 Physically Interacts with Mitoferrin-1 (Slc25a37) to Enhance Its Stability and Function in the Erythroid Mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 16263–16268. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Arimura, H.; Fukushige, T.; Minami, K.; Nishizawa, Y.; Tanimoto, A.; Kanekura, T.; Nakagawa, M.; Akiyama, S.-I.; Furukawa, T. Abcb10 Role in Heme Biosynthesis in Vivo: Abcb10 Knockout in Mice Causes Anemia with Protoporphyrin IX and Iron Accumulation. Mol. Cell. Biol. 2014, 34, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P. Tissue-Specific Regulation of Iron Metabolism and Heme Synthesis: Distinct Control Mechanisms in Erythroid Cells. Blood 1997, 89, 1–25. [Google Scholar] [Green Version]
- Napier, I.; Ponka, P.; Richardson, D.R. Iron Trafficking in the Mitochondrion: Novel Pathways Revealed by Disease. Blood 2005, 105, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Sachar, M.; Anderson, K.E.; Ma, X. Protoporphyrin IX: The Good, the Bad, and the Ugly. J. Pharmacol. Exp. Ther. 2016, 356, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Rich, P.R.; Maréchal, A. The Mitochondrial Respiratory Chain. Essays Biochem. 2010, 47, 1–23. [Google Scholar] [CrossRef]
- Cecchini, G. Function and Structure of Complex II of the Respiratory Chain. Annu. Rev. Biochem. 2003, 72, 77–109. [Google Scholar] [CrossRef] [Green Version]
- Fuss, J.O.; Tsai, C.-L.; Ishida, J.P.; Tainer, J.A. Emerging Critical Roles of Fe-S Clusters in DNA Replication and Repair. Biochim. Biophys. Acta 2015, 1853, 1253–1271. [Google Scholar] [CrossRef] [PubMed]
- Braymer, J.J.; Lill, R. Iron-Sulfur Cluster Biogenesis and Trafficking in Mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.M.; Cloonan, S.M. Mitochondrial Iron in Human Health and Disease. Annu. Rev. Physiol. 2019, 81, 453–482. [Google Scholar] [CrossRef] [PubMed]
- Land, T.; Rouault, T.A. Targeting of a Human Iron–Sulfur Cluster Assembly Enzyme, Nifs, to Different Subcellular Compartments Is Regulated through Alternative AUG Utilization. Mol. Cell 1998, 2, 807–815. [Google Scholar] [CrossRef]
- Rouault, T.A.; Maio, N. Biogenesis and Functions of Mammalian Iron-Sulfur Proteins in the Regulation of Iron Homeostasis and Pivotal Metabolic Pathways. J. Biol. Chem. 2017, 292, 12744–12753. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, T.L.; Lesuisse, E.; Pain, D.; Dancis, A. Frataxin and Mitochondrial FeS Cluster Biogenesis. J. Biol. Chem. 2010, 285, 26737–26743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxid. Redox Signal. 2018, 30, 1083–1095. [Google Scholar] [CrossRef]
- Tamir, S.; Paddock, M.L.; Darash-Yahana-Baram, M.; Holt, S.H.; Sohn, Y.S.; Agranat, L.; Michaeli, D.; Stofleth, J.T.; Lipper, C.H.; Morcos, F.; et al. Structure–Function Analysis of NEET Proteins Uncovers Their Role as Key Regulators of Iron and ROS Homeostasis in Health and Disease. SI FeS Proteins 2015, 1853, 1294–1315. [Google Scholar] [CrossRef]
- Ferecatu, I.; Gonçalves, S.; Golinelli-Cohen, M.-P.; Clémancey, M.; Martelli, A.; Riquier, S.; Guittet, E.; Latour, J.-M.; Puccio, H.; Drapier, J.-C.; et al. The Diabetes Drug Target MitoNEET Governs a Novel Trafficking Pathway to Rebuild an Fe-S Cluster into Cytosolic Aconitase/Iron Regulatory Protein 1. J. Biol. Chem. 2014, 289, 28070–28086. [Google Scholar] [CrossRef] [Green Version]
- Camponeschi, F.; Ciofi-Baffoni, S.; Banci, L. Anamorsin/Ndor1 Complex Reduces [2Fe–2S]-MitoNEET via a Transient Protein–Protein Interaction. J. Am. Chem. Soc. 2017, 139, 9479–9482. [Google Scholar] [CrossRef]
- Lipper, C.H.; Paddock, M.L.; Onuchic, J.N.; Mittler, R.; Nechushtai, R.; Jennings, P.A. Cancer-Related NEET Proteins Transfer 2Fe-2S Clusters to Anamorsin, a Protein Required for Cytosolic Iron-Sulfur Cluster Biogenesis. PLoS ONE 2015, 10, e0139699. [Google Scholar] [CrossRef] [PubMed]
- Landry, A.P.; Cheng, Z.; Ding, H. Reduction of Mitochondrial Protein MitoNEET [2Fe–2S] Clusters by Human Glutathione Reductase. Free Radic. Biol. Med. 2015, 81, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.E.; Crail, J.P.; Laffoon, M.M.; Fernandez, W.G.; Menze, M.A.; Konkle, M.E. Identification of Disulfide Bond Formation between MitoNEET and Glutamate Dehydrogenase 1. Biochemistry 2013, 52, 8969–8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karmi, O.; Holt, S.H.; Song, L.; Tamir, S.; Luo, Y.; Bai, F.; Adenwalla, A.; Darash-Yahana, M.; Sohn, Y.-S.; Jennings, P.A.; et al. Interactions between MitoNEET and NAF-1 in Cells. PLoS ONE 2017, 12, e0175796. [Google Scholar] [CrossRef] [PubMed]
- Lipper, C.H.; Karmi, O.; Sohn, Y.S.; Darash-Yahana, M.; Lammert, H.; Song, L.; Liu, A.; Mittler, R.; Nechushtai, R.; Onuchic, J.N.; et al. Structure of the Human Monomeric NEET Protein MiNT and Its Role in Regulating Iron and Reactive Oxygen Species in Cancer Cells. Proc. Natl. Acad. Sci. USA 2018, 115, 272. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, J.; Arosio, P.; Invernizzi, R.; Cazzola, M.; Volz, A.; Corsi, B.; Biasiotto, G.; Levi, S. Mitochondrial Ferritin: A New Player in Iron Metabolism. Blood Cells Mol. Dis. 2002, 29, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Guaraldo, M.; Santambrogio, P.; Rovelli, E.; Di Savino, A.; Saglio, G.; Cittaro, D.; Roetto, A.; Levi, S. Characterization of Human Mitochondrial Ferritin Promoter: Identification of Transcription Factors and Evidences of Epigenetic Control. Sci. Rep. 2016, 6, 33432. [Google Scholar] [CrossRef]
- Bystrom, L.M.; Rivella, S. Cancer Cells with Irons in the Fire. Free Radic. Biol. Med. 2015, 79, 337–342. [Google Scholar] [CrossRef]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and Cancer: Recent Insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef]
- Huang, X. Iron Overload and Its Association with Cancer Risk in Humans: Evidence for Iron as a Carcinogenic Metal. Met. Hum. Cancer 2003, 533, 153–171. [Google Scholar] [CrossRef]
- Kew, M.C. Hepatic Iron Overload and Hepatocellular Carcinoma. Liver Cancer 2014, 3, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.P.; Lee, J.H.; Tai, Y.-P.; Wen, C.; Wu, S.B.; Tsai, M.K.; Hsieh, D.P.H.; Chiang, H.-C.; Hsiung, C.A.; Hsu, C.Y.; et al. High Serum Iron Is Associated with Increased Cancer Risk. Cancer Res. 2014, 74, 6589. [Google Scholar] [CrossRef] [PubMed]
- Fracanzani, A.L.; Conte, D.; Fraquelli, M.; Taioli, E.; Mattioli, M.; Losco, A.; Fargion, S. Increased Cancer Risk in a Cohort of 230 Patients with Hereditary Hemochromatosis in Comparison to Matched Control Patients with Non–Iron-Related Chronic Liver Disease. Hepatology 2001, 33, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.D.; Coffman, L.G.; Chou, J.W.; Black, M.A.; Bergh, J.; D’Agostino, R., Jr.; Torti, S.V.; Torti, F.M. An Iron Regulatory Gene Signature Predicts Outcome in Breast Cancer. Cancer Res. 2011, 71, 6728–6737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A Ribonucleotide Reductase Gene Involved in a P53-Dependent Cell-Cycle Checkpoint for DNA Damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Thelander, L.; Gräslund, A.; Thelander, M. Continual Presence of Oxygen and Iron Required for Mammalian Ribonucleotide Reduction: Possible Regulation Mechanism. Biochem. Biophys. Res. Commun. 1983, 110, 859–865. [Google Scholar] [CrossRef]
- Terada, N.; Or, R.; Szepesi, A.; Lucas, J.J.; Gelfand, E.W. Definition of the Roles for Iron and Essential Fatty Acids in Cell Cycle Progression of Normal Human T Lymphocytes. Exp. Cell Res. 1993, 204, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Nurtjahja-Tjendraputra, E.; Fu, D.; Phang, J.M.; Richardson, D.R. Iron Chelation Regulates Cyclin D1 Expression via the Proteasome: A Link to Iron Deficiency–Mediated Growth Suppression. Blood 2007, 109, 4045. [Google Scholar] [CrossRef] [PubMed]
- Kulp, K.S.; Green, S.L.; Vulliet, P.R. Iron Deprivation Inhibits Cyclin-Dependent Kinase Activity and Decreases Cyclin D/CDK4 Protein Levels in Asynchronous MDA-MB-453 Human Breast Cancer Cells. Exp. Cell Res. 1996, 229, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Moussa, R.S.; Park, K.C.; Kovacevic, Z.; Richardson, D.R. Ironing out the Role of the Cyclin-Dependent Kinase Inhibitor, P21 in Cancer: Novel Iron Chelating Agents to Target P21 Expression and Activity. Iron Soul Life Earth Revisit. Chem. React. Ferroptosis Ther. 2019, 133, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, X. P53 Tumor Suppressor and Iron Homeostasis. FEBS J. 2019, 286, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P. The Regulation of Cyclin D1 Degradation: Roles in Cancer Development and the Potential for Therapeutic Invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Li, M.; Long, M.J.C.; Weiss, R.S. Ribonucleotide Reductase and Cancer: Biological Mechanisms and Targeted Therapies. Oncogene 2014, 34, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Puig, S.; Ramos-Alonso, L.; Romero, A.M.; Martínez-Pastor, M.T. The Elemental Role of Iron in DNA Synthesis and Repair. Metallomics 2017, 9, 1483–1500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. Essential Functions of Iron-Requiring Proteins in DNA Replication, Repair and Cell Cycle Control. Protein Cell 2014, 5, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Brosh, R.M., Jr. DNA Helicase and Helicase-Nuclease Enzymes with a Conserved Iron-Sulfur Cluster. Nucleic Acids Res. 2012, 40, 4247–4260. [Google Scholar] [CrossRef] [PubMed]
- Parish, J.L.; Rosa, J.; Wang, X.; Lahti, J.M.; Doxsey, S.J.; Androphy, E.J. The DNA Helicase ChlR1 Is Required for Sister Chromatid Cohesion in Mammalian Cells. J. Cell Sci. 2006, 119, 4857–4865. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.; Arab, N.T.T.; Greenwood, M.T. Iron Mediated Toxicity and Programmed Cell Death: A Review and a Re-Examination of Existing Paradigms. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2017, 1864, 399–430. [Google Scholar] [CrossRef]
- Yang, F.; Li, Y.; Yan, G.; Liu, T.; Feng, C.; Gong, R.; Yuan, Y.; Ding, F.; Zhang, L.; Idiiatullina, E.; et al. Inhibition of Iron Overload-Induced Apoptosis and Necrosis of Bone Marrow Mesenchymal Stem Cells by Melatonin. Oncotarget 2017, 8, 31626–31637. [Google Scholar] [CrossRef]
- Chen, M.; Cabantchik, Z.I.; Chan, S.; Chan, G.C.; Cheung, Y. Iron Overload and Apoptosis of HL-1 Cardiomyocytes: Effects of Calcium Channel Blockade. PLoS ONE 2014, 9, e112915. [Google Scholar] [CrossRef]
- Yu, Z.; Persson, H.L.; Eaton, J.W.; Brunk, U.T. Intralysosomal Iron: A Major Determinant of Oxidant-Induced Cell Death. Free Radic. Biol. Med. 2003, 34, 1243–1252. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T. Lysosomal Iron, Iron Chelation, and Cell Death. Antioxid. Redox Signal. 2012, 18, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.S.; von Schwarzenberg, K.; Lehr, T.; Ulrich, M.; Kubisch-Dohmen, R.; Liebl, J.; Trauner, D.; Menche, D.; Vollmar, A.M. Vacuolar-ATPase Inhibition Blocks Iron Metabolism to Mediate Therapeutic Effects in Breast Cancer. Cancer Res. 2015, 75, 2863–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin Receptor 1 in Cancer: A New Sight for Cancer Therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar] [PubMed]
- O’Donnell, K.A.; Yu, D.; Zeller, K.I.; Kim, J.-W.; Racke, F.; Thomas-Tikhonenko, A.; Dang, C.V. Transferrin Receptor 1, a Direct c-Myc Target, Is Necessary for Cell-Cycle Progression and Stimulates Cellular Proliferation under Limiting Conditions. Cancer Res. 2005, 65 (Suppl. S9), 6119. [Google Scholar]
- O’Donnell, K.A.; Yu, D.; Zeller, K.I.; Kim, J.-W.; Racke, F.; Thomas-Tikhonenko, A.; Dang, C.V. Activation of Transferrin Receptor 1 by C-Myc Enhances Cellular Proliferation and Tumorigenesis. Mol. Cell. Biol. 2006, 26, 2373–2386. [Google Scholar] [CrossRef]
- Röhrs, S.; Kutzner, N.; Vlad, A.; Grunwald, T.; Ziegler, S.; Müller, O. Chronological Expression of Wnt Target Genes Ccnd1, Myc, Cdkn1a, Tfrc, Plf1 and Ramp3. Cell Biol. Int. 2009, 33, 501–508. [Google Scholar] [CrossRef]
- Shah, Y.M.; Xie, L. Hypoxia-Inducible Factors Link Iron Homeostasis and Erythropoiesis. Gastroenterology 2014, 146, 630–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Tapryal, N.; Mukherjee, R.; Kumar, R.; Mukhopadhyay, C.K. Insulin Promotes Iron Uptake in Human Hepatic Cell by Regulating Transferrin Receptor-1 Transcription Mediated by Hypoxia Inducible Factor-1. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2013, 1832, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Lok, C.N.; Ponka, P. Identification of a Hypoxia Response Element in the Transferrin Receptor Gene. J. Biol. Chem. 1999, 274, 24147–24152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Whitton, B.; Okamoto, H.; Packham, G.; Crabb, S.J. Vacuolar ATPase as a Potential Therapeutic Target and Mediator of Treatment Resistance in Cancer. Cancer Med. 2018, 7, 3800–3811. [Google Scholar] [CrossRef] [PubMed]
- Lambe, T.; Simpson, R.J.; Dawson, S.; Bouriez-Jones, T.; Crockford, T.L.; Lepherd, M.; Latunde-Dada, G.O.; Robinson, H.; Raja, K.B.; Campagna, D.R.; et al. Identification of a Steap3 Endosomal Targeting Motif Essential for Normal Iron Metabolism. Blood 2009, 113, 1805–1808. [Google Scholar] [CrossRef]
- Gomes, I.M.; Maia, C.J.; Santos, C.R. STEAP Proteins: From Structure to Applications in Cancer Therapy. Mol. Cancer Res. 2012, 10, 573. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap Proteins Are Metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef]
- Isobe, T.; Baba, E.; Arita, S.; Komoda, M.; Tamura, S.; Shirakawa, T.; Ariyama, H.; Takaishi, S.; Kusaba, H.; Ueki, T.; et al. Human STEAP3 Maintains Tumor Growth under Hypoferric Condition. Exp. Cell Res. 2011, 317, 2582–2591. [Google Scholar] [CrossRef]
- Ramey, G.; Deschemin, J.-C.; Durel, B.; Canonne-Hergaux, F.; Nicolas, G.; Vaulont, S. Hepcidin Targets Ferroportin for Degradation in Hepatocytes. Haematologica 2010, 95, 501–504. [Google Scholar] [CrossRef]
- Vela, D.; Vela-Gaxha, Z. Differential Regulation of Hepcidin in Cancer and Non-Cancer Tissues and Its Clinical Implications. Exp. Mol. Med. 2018, 50, e436. [Google Scholar] [CrossRef] [PubMed]
- Toshiyama, R.; Konno, M.; Eguchi, H.; Asai, A.; Noda, T.; Koseki, J.; Asukai, K.; Ohashi, T.; Matsushita, K.; Iwagami, Y.; et al. Association of Iron Metabolic Enzyme Hepcidin Expression Levels with the Prognosis of Patients with Pancreatic Cancer. Oncol. Lett. 2018, 15, 8125–8133. [Google Scholar] [CrossRef] [PubMed]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and Iron Regulation in Breast Cancer Progression and Prognosis. Sci. Transl. Med. 2010, 2, 43–56. [Google Scholar] [CrossRef]
- Zhao, B.; Li, R.; Cheng, G.; Li, Z.; Zhang, Z.; Li, J.; Zhang, G.; Bi, C.; Hu, C.; Yang, L.; et al. Role of Hepcidin and Iron Metabolism in the Onset of Prostate Cancer. Oncol. Lett. 2018, 15, 9953–9958. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current Questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.-I.; Schwartz, J.M.; Maldonado, E.N.; Lemasters, J.J.; Nieminen, A.-L. Mitoferrin-2-Dependent Mitochondrial Iron Uptake Sensitizes Human Head and Neck Squamous Carcinoma Cells to Photodynamic Therapy. J. Biol. Chem. 2013, 288, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Nie, G.; Chen, G.; Sheftel, A.D.; Pantopoulos, K.; Ponka, P. In Vivo Tumor Growth Is Inhibited by Cytosolic Iron Deprivation Caused by the Expression of Mitochondrial Ferritin. Blood 2006, 108, 2428–2434. [Google Scholar] [CrossRef]
- Lu, Z.; Nie, G.; Li, Y.; Soe-lin, S.; Tao, Y.; Cao, Y.; Zhang, Z.; Liu, N.; Ponka, P.; Zhao, B. Overexpression of Mitochondrial Ferritin Sensitizes Cells to Oxidative Stress Via an Iron-Mediated Mechanism. Antioxid. Redox Signal. 2009, 11, 1791–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierich, A.; Gansmuller, A.; Koutnikova, H.; Puccio, H.; Fischbeck, K.; LeMeur, M.; Kœnig, M.; Cossée, M.; Dollé, P. Inactivation of the Friedreich Ataxia Mouse Gene Leads to Early Embryonic Lethality without Iron Accumulation. Hum. Mol. Genet. 2000, 9, 1219–1226. [Google Scholar] [CrossRef]
- Houten, B.V.; Karthikeyan, G.; Isaya, G.; Santos, J.H.; Graziewicz, M.A.; Resnick, M.A.; Copeland, W.C. Reduction in Frataxin Causes Progressive Accumulation of Mitochondrial Damage. Hum. Mol. Genet. 2003, 12, 3331–3342. [Google Scholar] [CrossRef]
- Guccini, I.; Serio, D.; Condò, I.; Rufini, A.; Tomassini, B.; Mangiola, A.; Maira, G.; Anile, C.; Fina, D.; Pallone, F.; et al. Frataxin Participates to the Hypoxia-Induced Response in Tumors. Cell Death Dis. 2011, 2, e123. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.; Thierbach, R.; Voigt, A.; Drewes, G.; Mietzner, B.; Steinberg, P.; Pfeiffer, A.; Ristow, M. Induction of Oxidative Metabolism by Mitochondrial Frataxin Inhibits Cancer Growth: Otto Warburg Revisited. J. Biol. Chem. 2006, 281, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, R.; Lan, N.N.; Tai, T.T.; Adachi, Y.; Kawazoe, A.; Mu, A.; Taketani, S. P53 Directly Regulates the Transcription of the Human Frataxin Gene and Its Lack of Regulation in Tumor Cells Decreases the Utilization of Mitochondrial Iron. Gene 2014, 551, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.F.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Mitochondrial Biogenesis in Epithelial Cancer Cells Promotes Breast Cancer Tumor Growth and Confers Autophagy Resistance. Cell Cycle 2012, 11, 4174–4180. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.-S.; Tamir, S.; Song, L.; Michaeli, D.; Matouk, I.; Conlan, A.R.; Harir, Y.; Holt, S.H.; Shulaev, V.; Paddock, M.L.; et al. NAF-1 and MitoNEET Are Central to Human Breast Cancer Proliferation by Maintaining Mitochondrial Homeostasis and Promoting Tumor Growth. Proc. Natl. Acad. Sci. USA 2013, 110, 14676–14681. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.H.; Darash-Yahana, M.; Sohn, Y.S.; Song, L.; Karmi, O.; Tamir, S.; Michaeli, D.; Luo, Y.; Paddock, M.L.; Jennings, P.A.; et al. Activation of Apoptosis in NAF-1-Deficient Human Epithelial Breast Cancer Cells. J. Cell Sci. 2016, 129, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Shen, S.; Wu, J.; Hua, Y.; Kuang, M.; Li, S.; Peng, B. CISD2 Associated with Proliferation Indicates Negative Prognosis in Patients with Hepatocellular Carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 13725–13738. [Google Scholar]
- Darash-Yahana, M.; Pozniak, Y.; Lu, M.; Sohn, Y.-S.; Karmi, O.; Tamir, S.; Bai, F.; Song, L.; Jennings, P.A.; Pikarsky, E.; et al. Breast Cancer Tumorigenicity Is Dependent on High Expression Levels of NAF-1 and the Lability of Its Fe-S Clusters. Proc. Natl. Acad. Sci. USA 2016, 113, 10890–10895. [Google Scholar] [CrossRef]
- Liu, L.; Xia, M.; Wang, J.; Zhang, W.; Zhang, Y.; He, M. CISD2 Expression Is a Novel Marker Correlating with Pelvic Lymph Node Metastasis and Prognosis in Patients with Early-Stage Cervical Cancer. Med. Oncol. 2014, 31, 183. [Google Scholar] [CrossRef]
- Hooda, J.; Cadinu, D.; Alam, M.M.; Shah, A.; Cao, T.M.; Sullivan, L.A.; Brekken, R.; Zhang, L. Enhanced Heme Function and Mitochondrial Respiration Promote the Progression of Lung Cancer Cells. PLoS ONE 2013, 8, e63402. [Google Scholar] [CrossRef]
- Sohoni, S.; Ghosh, P.; Wang, T.; Kalainayakan, S.P.; Vidal, C.; Dey, S.; Konduri, P.C.; Zhang, L. Elevated Heme Synthesis and Uptake Underpin Intensified Oxidative Metabolism and Tumorigenic Functions in Non-Small Cell Lung Cancer Cells. Cancer Res. 2019, 79, 2511–2525. [Google Scholar] [CrossRef] [PubMed]
- Hooda, J.; Alam, M.M.; Zhang, L. Evaluating the Association of Heme and Heme Metabolites with Lung Cancer Bioenergetics and Progression. Metabolomics 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, T.; Otsuka, S.; Kobayashi, T.; Okajima, H.; Matsumoto, K.; Hagiya, Y.; Inoue, K.; Shuin, T.; Nakajima, M.; Tanaka, T.; et al. Dormant Cancer Cells Accumulate High Protoporphyrin IX Levels and Are Sensitive to 5-Aminolevulinic Acid-Based Photodynamic Therapy. Sci. Rep. 2016, 6, 36478. [Google Scholar] [CrossRef] [PubMed]
- Kemmner, W.; Wan, K.; Rüttinger, S.; Ebert, B.; Macdonald, R.; Klamm, U.; Moesta, K.T. Silencing of Human Ferrochelatase Causes Abundant Protoporphyrin-IX Accumulation in Colon Cancer. FASEB J. 2007, 22, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Spitz, D.R. Manipulations of Redox Metabolism for Enhancing Radiation Therapy Responses: A Historical Perspective and Novel Hypothesis. Target. Redox Metab. Enhancing Radiat. Ther. Responses 2019, 29, 1–5. [Google Scholar] [CrossRef]
- Zhu, Y.; Dean, A.E.; Horikoshi, N.; Heer, C.; Spitz, D.R.; Gius, D. Emerging Evidence for Targeting Mitochondrial Metabolic Dysfunction in Cancer Therapy. J. Clin. Investig. 2018, 128, 3682–3691. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W.; Buettner, G.R. Role of Superoxide Dismutase in Cancer: A Review. Cancer Res. 1979, 39, 1141–1149. [Google Scholar]
- Oberley, L.W.; Oberley, T.D.; Buettner, G.R. Cell Differentation, Aging and Cancer: The Possible Roles of Superoxide and Superoxide Dismutases. Med. Hypotheses 1980, 6, 249–268. [Google Scholar] [CrossRef]
- Oberley, L.W.; Oberley, T.D.; Buettner, G.R. Cell Division in Normal and Transformed Cells: The Possible Role of Superoxide and Hydrogen Peroxide. Med. Hypotheses 1981, 7, 21–42. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Tuttle, S.W.; Varnes, M.E.; Mitchell, J.B.; Biaglow, J.E. Sensitivity to Chemical Oxidants and Radiation in CHO Cell Lines Deficient in Oxidative Pentose Cycle Activity. Int. J. Radiat. Oncol. Biol. Phys. 1992, 22, 671–675. [Google Scholar] [CrossRef]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased Levels of Superoxide and H2O2 Mediate the Differential Susceptibility of Cancer Cells versus Normal Cells to Glucose Deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, α-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.; Anne Jurkiewicz, B. Catalytic Metals, Ascorbate and Free Radicals: Combinations to Avoid. Radiat. Res. 1996, 145. [Google Scholar] [CrossRef]
- Wu, T.; Li, Y.; Liu, B.; Zhang, S.; Wu, L.; Zhu, X.; Chen, Q. Expression of Ferritin Light Chain (FTL) Is Elevated in Glioblastoma, and FTL Silencing Inhibits Glioblastoma Cell Proliferation via the GADD45/JNK Pathway. PLoS ONE 2016, 11, e0149361. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; Miller, T.E.; Wu, Q.; Flavahan, W.A.; Das, N.K.; Hale, J.S.; Hubert, C.G.; Mack, S.C.; Jarrar, A.M.; Karl, R.T.; et al. Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell 2015, 28, 441–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldi, A.; Lombardi, D.; Russo, P.; Palescandolo, E.; De Luca, A.; Santini, D.; Baldi, F.; Rossiello, L.; Dell’Anna, M.L.; Mastrofrancesco, A.; et al. Ferritin Contributes to Melanoma Progression by Modulating Cell Growth and Sensitivity to Oxidative Stress. Clin. Cancer Res. 2005, 11, 3175–3183. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.C.; Wang, F.; Elliott, R.L.; Head, J.F. Expression of Transferrin Receptor and Ferritin H-Chain MRNA Are Associated with Clinical and Histopathological Prognostic Indicators in Breast Cancer. Anticancer Res. 2001, 21, 541–549. [Google Scholar]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Chiu, W.-T.; Huang, Y.-F.; Tsai, H.-Y.; Chen, C.-C.; Chang, C.-H.; Huang, S.-C.; Hsu, K.-F.; Chou, C.-Y. FOXM1 Confers to Epithelial-Mesenchymal Transition, Stemness and Chemoresistance in Epithelial Ovarian Carcinoma Cells. Oncotarget 2014, 6, 2349–2365. [Google Scholar] [CrossRef]
- Yu, C.-P.; Yu, S.; Shi, L.; Wang, S.; Li, Z.-X.; Wang, Y.-H.; Sun, C.-J.; Liang, J. FoxM1 Promotes Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma by Targeting Snai1. Mol. Med. Rep. 2017, 16, 5181–5188. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, H.; Tan, G.; Gao, W.; Cheng, L.; Jiang, X.; Yu, L.; Tan, Y. FOXM1 Promotes the Epithelial to Mesenchymal Transition by Stimulating the Transcription of Slug in Human Breast Cancer. Cancer Lett. 2013, 340, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Niu, Y.; Huang, C. Role of FoxM1 in the Progression and Epithelial to Mesenchymal Transition of Gastrointestinal Cancer. Recent Pat. Anticancer Drug Discov. 2017, 12, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Becton, D.L.; Bryles, P. Deferoxamine Inhibition of Human Neuroblastoma Viability and Proliferation. Cancer Res. 1988, 48 Pt 1, 7189–7192. [Google Scholar]
- Simonart, T.; Boelaert, J.R.; Mosselmans, R.; Andrei, G.; Noel, J.-C.; De Clercq, E.; Snoeck, R. Antiproliferative and Apoptotic Effects of Iron Chelators on Human Cervical Carcinoma Cells. Gynecol. Oncol. 2002, 85, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Becton, D.L.; Roberts, B. Antileukemic Effects of Deferoxamine on Human Myeloid Leukemia Cell Lines. Cancer Res. 1989, 49, 4809–4812. [Google Scholar] [PubMed]
- Brard, L.; Granai, C.O.; Swamy, N. Iron Chelators Deferoxamine and Diethylenetriamine Pentaacetic Acid Induce Apoptosis in Ovarian Carcinoma. Gynecol. Oncol. 2006, 100, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Donfrancesco, A.; Deb, G.; Dominici, C.; Pileggi, D.; Castello, M.A.; Helson, L. Effects of a Single Course of Deferoxamine in Neuroblastoma Patients. Cancer Res. 1990, 50, 4929. [Google Scholar] [PubMed]
- Donfrancesco, A.; De Bernardi, B.; Carli, M.; Mancini, A.; Nigro, M.; De Sio, L.; Casale, F.; Bagnulo, S.; Helson, L.; Deb, G. Deferoxamine Followed by Cyclophosphamide, Etoposide, Carboplatin, Thiotepa, Induction Regimen in Advanced Neuroblastoma: Preliminary Results. Genet. Cell. Biol. Clin. Manag. Hum. Neuroblastoma 1995, 31, 612–615. [Google Scholar] [CrossRef]
- Yamasaki, T.; Terai, S.; Sakaida, I. Deferoxamine for Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2011, 365, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Blatt, J. Deferoxamine in Children with Recurrent Neuroblastoma. Anticancer Res. 1994, 14, 2109–2112. [Google Scholar]
- Simões, R.V.; Veeraperumal, S.; Serganova, I.S.; Kruchevsky, N.; Varshavsky, J.; Blasberg, R.G.; Ackerstaff, E.; Koutcher, J.A. Inhibition of Prostate Cancer Proliferation by Deferiprone. NMR Biomed. 2017, 30, e3712. [Google Scholar] [CrossRef] [PubMed]
- Tury, S.; Assayag, F.; Bonin, F.; Chateau-Joubert, S.; Servely, J.-L.; Vacher, S.; Becette, V.; Caly, M.; Rapinat, A.; Gentien, D.; et al. The Iron Chelator Deferasirox Synergises with Chemotherapy to Treat Triple-Negative Breast Cancers. J. Pathol. 2018, 246, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic Acid: Chemistry, Biology and the Treatment of Cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.; Campbell, A. The Orthomolecular Treatment of Cancer II. Clinical Trial of High-Dose Ascorbic Acid Supplements in Advanced Human Cancer. Chem. Biol. Interact. 1974, 9, 285–315. [Google Scholar] [CrossRef]
- Cameron, E.; Campbell, A.; Jack, T. The Orthomolecular Treatment of Cancer: III. Reticulum Cell Sarcoma: Double Complete Regression Induced by High-Dose Ascorbic Acid Therapy. Chem. Biol. Interact. 1975, 11, 387–393. [Google Scholar] [CrossRef]
- Cameron, E.; Pauling, L. Supplemental Ascorbate in the Supportive Treatment of Cancer: Prolongation of Survival Times in Terminal Human Cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689. [Google Scholar] [CrossRef]
- Cameron, E.; Pauling, L. Supplemental Ascorbate in the Supportive Treatment of Cancer: Reevaluation of Prolongation of Survival Times in Terminal Human Cancer. Proc. Natl. Acad. Sci. USA 1978, 75, 4538–4542. [Google Scholar] [CrossRef]
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of High-Dose Vitamin C (Ascorbic Acid) Therapy to Benefit Patients with Advanced Cancer. N. Engl. J. Med. 1979, 301, 687–690. [Google Scholar] [CrossRef]
- Graumlich, J.F.; Ludden, T.M.; Conry-Cantilena, C.; Cantilena, L.R.; Wang, Y.; Levine, M. Pharmacokinetic Model of Ascorbic Acid in Healthy Male Volunteers During Depletion and Repletion. Pharm. Res. 1997, 14, 1133–1139. [Google Scholar] [CrossRef]
- Riordan, H.D.; Riordan, N.; Jackson, J.A.; Casciari, J.; Hunninghake, R.; Gonzalez, M.; Mora, E.; Miranda-Massari, J.; Rosario, N.; Rivera, A. Intravenous Vitamin C as a Chemotherapy Agent: A Report on Clinical Cases. Puerto Rico Health Sci. J. 2004, 23, 115–118. [Google Scholar]
- Monti, D.A.; Mitchell, E.; Bazzan, A.J.; Littman, S.; Zabrecky, G.; Yeo, C.J.; Pillai, M.V.; Newberg, A.B.; Deshmukh, S.; Levine, M. Phase I Evaluation of Intravenous Ascorbic Acid in Combination with Gemcitabine and Erlotinib in Patients with Metastatic Pancreatic Cancer. PLoS ONE 2012, 7, e29794. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Roberts, L.J., 2nd; et al. Pharmacological Ascorbate with Gemcitabine for the Control of Metastatic and Node-Positive Pancreatic Cancer (PACMAN): Results from a Phase I Clinical Trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Couple | Eo’ (mV) |
|---|---|
| Fe3+-transferrin/ Fe2+-transferrin | −400 (pH = 7.3) |
| Fe3+-ferritin/ Fe2+-ferritin | −190 |
| O2/ O2•− | −160 (−330) a |
| Fe3+-DETAPAC/ Fe2+-DETAPAC | 30 |
| Fe3+-citrate/ Fe2+-citrate | 100 |
| Fe3+-ADP/ Fe2+-ADP | 100 |
| Fe3+/Fe2+ (aqueous) | 110 |
| Fe3+-EDTA/Fe2+-EDTA | 120 |
| H2O2, H+/H2O, HO• | 320 mV |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petronek, M.S.; Spitz, D.R.; Buettner, G.R.; Allen, B.G. Linking Cancer Metabolic Dysfunction and Genetic Instability through the Lens of Iron Metabolism. Cancers 2019, 11, 1077. https://doi.org/10.3390/cancers11081077
Petronek MS, Spitz DR, Buettner GR, Allen BG. Linking Cancer Metabolic Dysfunction and Genetic Instability through the Lens of Iron Metabolism. Cancers. 2019; 11(8):1077. https://doi.org/10.3390/cancers11081077
Chicago/Turabian StylePetronek, Michael S., Douglas R. Spitz, Garry R. Buettner, and Bryan G. Allen. 2019. "Linking Cancer Metabolic Dysfunction and Genetic Instability through the Lens of Iron Metabolism" Cancers 11, no. 8: 1077. https://doi.org/10.3390/cancers11081077
APA StylePetronek, M. S., Spitz, D. R., Buettner, G. R., & Allen, B. G. (2019). Linking Cancer Metabolic Dysfunction and Genetic Instability through the Lens of Iron Metabolism. Cancers, 11(8), 1077. https://doi.org/10.3390/cancers11081077