PTEN Tumor-Suppressor: The Dam of Stemness in Cancer

 , , and
, , and
Abstract
:1. Introduction
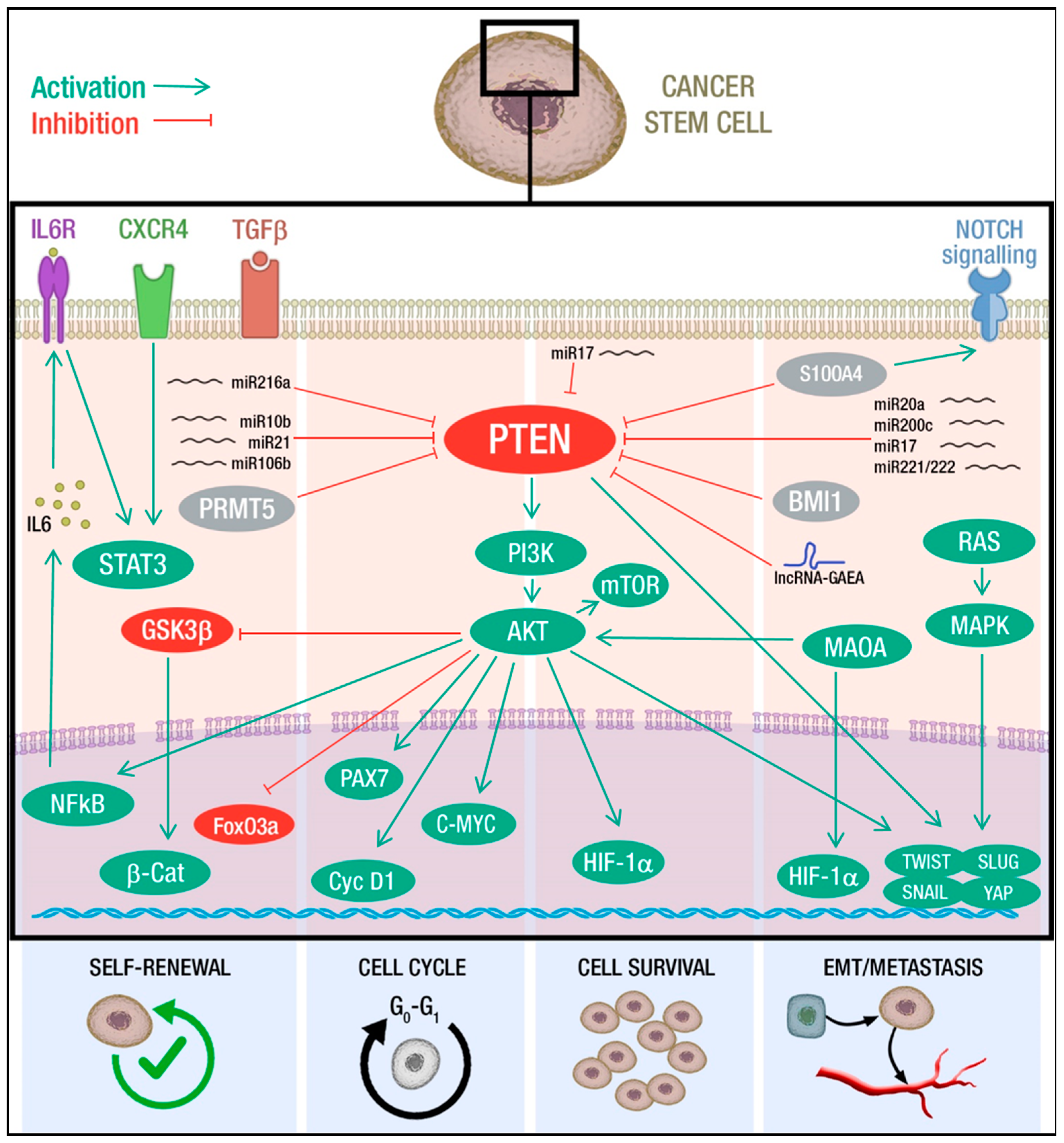
2. PTEN-Mediated Control of CSCs Hallmarks
2.1. PTEN and Self-Renewal
2.2. PTEN and Cell Cycle
2.3. PTEN and Cell Survival
2.4. PTEN and EMT-Metastasis
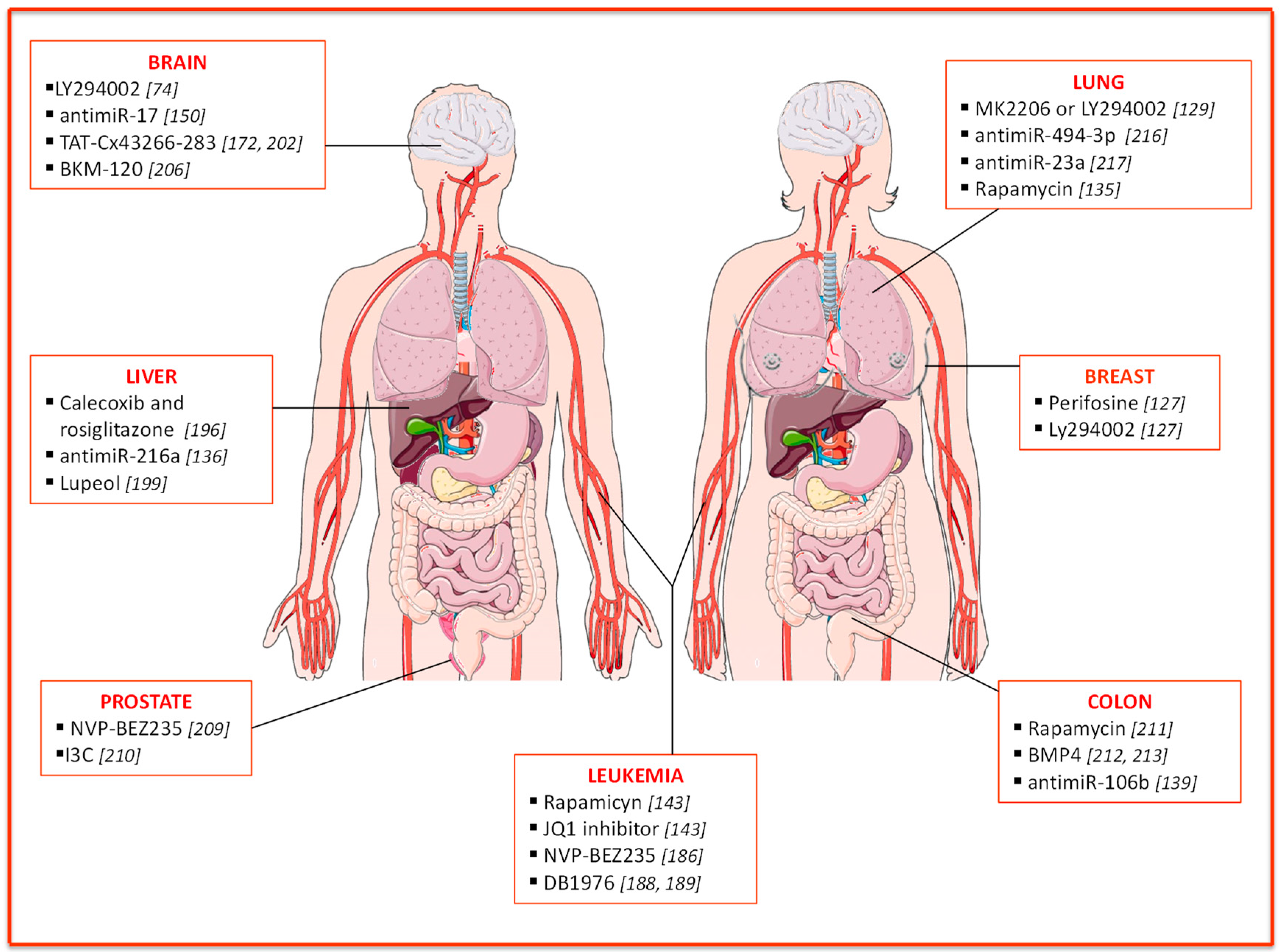
3. PTEN Implication in Therapy Resistance
3.1. Breast Cancer
3.2. Leukemia
3.3. Liver Cancer
3.4. Brain Cancer
3.5. Prostate Cancer
3.6. Colon Cancer
3.7. Lung Cancer
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Li, D.M.; Sun, H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997, 57, 2124–2129. [Google Scholar] [PubMed]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofano, A.; Pandolfi, P.P. The multiple roles of PTEN in tumor suppression. Cell 2000, 100, 387–390. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Niki, M.; Dotan, Z.A.; Koutcher, J.A.; Di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; Van Dyke, T.; et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003, 1, e59. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofano, A.; Kotsi, P.; Peng, Y.F.; Cordon-Cardo, C.; Elkon, K.B.; Pandolfi, P.P. Impaired Fas response and autoimmunity in Pten+/- mice. Science 1999, 285, 2122–2125. [Google Scholar] [CrossRef]
- Berger, A.H.; Knudson, A.G.; Pandolfi, P.P. A continuum model for tumour suppression. Nature 2011, 476, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; de la Pompa, J.L.; Stambolic, V.; Elia, A.J.; Sasaki, T.; del Barco Barrantes, I.; Ho, A.; Wakeham, A.; Itie, A.; Khoo, W.; et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr. Biol. 1998, 8, 1169–1178. [Google Scholar] [CrossRef] [Green Version]
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Podsypanina, K.; Ellenson, L.H.; Nemes, A.; Gu, J.; Tamura, M.; Yamada, K.M.; Cordon-Cardo, C.; Catoretti, G.; Fisher, P.E.; Parsons, R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc. Natl. Acad. Sci. USA 1999, 96, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Ngeow, J.; Sesock, K.; Eng, C. Clinical Implications for Germline PTEN Spectrum Disorders. Endocrinol. Metab. Clin. North. Am. 2017, 46, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Karikomi, M.; Naidu, S.; Rajmohan, R.; Caserta, E.; Chen, H.Z.; Rawahneh, M.; Moffitt, J.; Stephens, J.A.; Fernandez, S.A.; et al. Allele-specific tumor spectrum in pten knockin mice. Proc. Natl. Acad. Sci. USA 2010, 107, 5142–5147. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef]
- Khan, S.; Kumagai, T.; Vora, J.; Bose, N.; Sehgal, I.; Koeffler, P.H.; Bose, S. PTEN promoter is methylated in a proportion of invasive breast cancers. Int. J. Cancer 2004, 112, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006, 66, 6546–6552. [Google Scholar] [CrossRef]
- Salvesen, H.B.; MacDonald, N.; Ryan, A.; Jacobs, I.J.; Lynch, E.D.; Akslen, L.A.; Das, S. PTEN methylation is associated with advanced stage and microsatellite instability in endometrial carcinoma. Int. J. Cancer 2001, 91, 22–26. [Google Scholar] [CrossRef]
- Soria, J.C.; Lee, H.Y.; Lee, J.I.; Wang, L.; Issa, J.P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 2002, 8, 1178–1184. [Google Scholar]
- Zhang, J.; Lee, Y.R.; Dang, F.; Gan, W.; Menon, A.V.; Katon, J.M.; Hsu, C.H.; Asara, J.M.; Tibarewal, P.; Leslie, N.R.; et al. PTEN Methylation by NSD2 Controls Cellular Sensitivity to DNA Damage. Cancer Discov. 2019. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.G.; Wang, J.J.; Zhao, F.; Liu, Q.; Jiang, K.; Yang, G.H. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin. Chim. Acta 2010, 411, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Ciuffreda, L.; Di Sanza, C.; Cesta Incani, U.; Eramo, A.; Desideri, M.; Biagioni, F.; Passeri, D.; Falcone, I.; Sette, G.; Bergamo, P.; et al. The mitogen-activated protein kinase (MAPK) cascade controls phosphatase and tensin homolog (PTEN) expression through multiple mechanisms. J. Mol. Med. 2012, 90, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yu, D. PI(3)king apart PTEN’s role in cancer. Clin. Cancer Res. 2010, 16, 4325–4330. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Leevers, S.J.; Vanhaesebroeck, B.; Waterfield, M.D. Signalling through phosphoinositide 3-kinases: The lipids take centre stage. Curr. Opin. Cell Biol. 1999, 11, 219–225. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.P.; Stolarov, J.P.; Eng, C.; Li, J.; Wang, S.I.; Wigler, M.H.; Parsons, R.; Tonks, N.K. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. USA 1997, 94, 9052–9057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.C.; Piccini, A.; Myers, M.P.; Van Aelst, L.; Tonks, N.K. Functional analysis of the protein phosphatase activity of PTEN. Biochem. J. 2012, 444, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Tibarewal, P.; Zilidis, G.; Spinelli, L.; Schurch, N.; Maccario, H.; Gray, A.; Perera, N.M.; Davidson, L.; Barton, G.J.; Leslie, N.R. PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Sci. Signal. 2012, 5, ra18. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.M.; Ji, Y.; Zhang, C.; Dong, S.S.; Yang, S.; Xiong, Z.; Ge, M.K.; Yu, Y.; Xia, L.; Guo, M.; et al. Nuclear PTEN safeguards pre-mRNA splicing to link Golgi apparatus for its tumor suppressive role. Nat. Commun. 2018, 9, 2392. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Zhao, M.; Depinho, R.A.; Furnari, F.B.; Cavenee, W.K. Cellular transformation by the MSP58 oncogene is inhibited by its physical interaction with the PTEN tumor suppressor. Proc. Natl. Acad. Sci. USA 2005, 102, 2703–2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchon, S.M.; Waite, K.A.; Eng, C. The nuclear affairs of PTEN. J. Cell Sci 2008, 121, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimm, O.; Perren, A.; Weng, L.P.; Marsh, D.J.; Yeh, J.J.; Ziebold, U.; Gil, E.; Hinze, R.; Delbridge, L.; Lees, J.A.; et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am. J. Pathol. 2000, 156, 1693–1700. [Google Scholar] [CrossRef]
- Ginn-Pease, M.E.; Eng, C. Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G0-G1 in MCF-7 cells. Cancer Res. 2003, 63, 282–286. [Google Scholar] [PubMed]
- Sano, T.; Lin, H.; Chen, X.; Langford, L.A.; Koul, D.; Bondy, M.L.; Hess, K.R.; Myers, J.N.; Hong, Y.K.; Yung, W.K.; et al. Differential expression of MMAC/PTEN in glioblastoma multiforme: Relationship to localization and prognosis. Cancer Res. 1999, 59, 1820–1824. [Google Scholar] [PubMed]
- Lachyankar, M.B.; Sultana, N.; Schonhoff, C.M.; Mitra, P.; Poluha, W.; Lambert, S.; Quesenberry, P.J.; Litofsky, N.S.; Recht, L.D.; Nabi, R.; et al. A role for nuclear PTEN in neuronal differentiation. J. Neurosci. 2000, 20, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.C.; Zhou, X.P.; Cummings, M.C.; Pavey, S.; Hayward, N.K.; Eng, C. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. Int. J. Cancer 2002, 99, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Putz, U.; Howitt, J.; Doan, A.; Goh, C.P.; Low, L.H.; Silke, J.; Tan, S.S. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal. 2012, 5, ra70. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 2013, 341, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; He, S.; Yang, J.; Jia, X.; Wang, P.; Chen, X.; Zhang, Z.; Zou, X.; McNutt, M.A.; Shen, W.H.; et al. PTENalpha, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 2014, 19, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Chen, X.; Yin, Q.; Ruan, D.; Zhao, X.; Zhang, C.; McNutt, M.A.; Yin, Y. PTENbeta is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat. Commun. 2017, 8, 14771. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Falcone, I.; Incani, U.C.; Del Curatolo, A.; Conciatori, F.; Matteoni, S.; Vari, S.; Vaccaro, V.; Cognetti, F.; Milella, M. PTEN expression and function in adult cancer stem cells and prospects for therapeutic targeting. Adv. Biol. Regul. 2014, 56, 66–80. [Google Scholar] [CrossRef]
- Lavictoire, S.J.; Gont, A.; Julian, L.M.; Stanford, W.L.; Vlasschaert, C.; Gray, D.A.; Jomaa, D.; Lorimer, I.A.J. Engineering PTEN-L for Cell-Mediated Delivery. Mol. Ther. Methods Clin. Dev. 2018, 9, 12–22. [Google Scholar] [CrossRef]
- Jiang, B.H.; Liu, L.Z. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv. Cancer Res. 2009, 102, 19–65. [Google Scholar] [CrossRef]
- Sizemore, G.M.; Balakrishnan, S.; Hammer, A.M.; Thies, K.A.; Trimboli, A.J.; Wallace, J.A.; Sizemore, S.T.; Kladney, R.D.; Woelke, S.A.; Yu, L.; et al. Stromal PTEN inhibits the expansion of mammary epithelial stem cells through Jagged-1. Oncogene 2017, 36, 2297. [Google Scholar] [CrossRef]
- Luo, X.; Liao, R.; Hanley, K.L.; Zhu, H.H.; Malo, K.N.; Hernandez, C.; Wei, X.; Varki, N.M.; Alderson, N.; Chu, C.; et al. Dual Shp2 and Pten Deficiencies Promote Non-alcoholic Steatohepatitis and Genesis of Liver Tumor-Initiating Cells. Cell Rep. 2016, 17, 2979–2993. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef]
- Shojaee, S.; Chan, L.N.; Buchner, M.; Cazzaniga, V.; Cosgun, K.N.; Geng, H.; Qiu, Y.H.; von Minden, M.D.; Ernst, T.; Hochhaus, A.; et al. PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat. Med. 2016, 22, 379–387. [Google Scholar] [CrossRef]
- Li, Y.; Guessous, F.; Kwon, S.; Kumar, M.; Ibidapo, O.; Fuller, L.; Johnson, E.; Lal, B.; Hussaini, I.; Bao, Y.; et al. PTEN has tumor-promoting properties in the setting of gain-of-function p53 mutations. Cancer Res. 2008, 68, 1723–1731. [Google Scholar] [CrossRef]
- Costa, H.A.; Leitner, M.G.; Sos, M.L.; Mavrantoni, A.; Rychkova, A.; Johnson, J.R.; Newton, B.W.; Yee, M.C.; De La Vega, F.M.; Ford, J.M.; et al. Discovery and functional characterization of a neomorphic PTEN mutation. Proc. Natl. Acad. Sci. USA 2015, 112, 13976–13981. [Google Scholar] [CrossRef] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Manic, G.; Sistigu, A.; Corradi, F.; Musella, M.; De Maria, R.; Vitale, I. Replication stress response in cancer stem cells as a target for chemotherapy. Semin. Cancer Biol. 2018, 53, 31–41. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—what challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Vermeulen, L.; Todaro, M.; de Sousa Mello, F.; Sprick, M.R.; Kemper, K.; Perez Alea, M.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Fiori, M.E.; Villanova, L.; De Maria, R. Cancer stem cells: At the forefront of personalized medicine and immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 1–11. [Google Scholar] [CrossRef]
- Zeuner, A.; Todaro, M.; Stassi, G.; De Maria, R. Colorectal cancer stem cells: From the crypt to the clinic. Cell Stem Cell 2014, 15, 692–705. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef]
- Koury, J.; Zhong, L.; Hao, J. Targeting Signaling Pathways in Cancer Stem Cells for Cancer Treatment. Stem Cells Int. 2017, 2017, 2925869. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef]
- Pourmand, G.; Ziaee, A.A.; Abedi, A.R.; Mehrsai, A.; Alavi, H.A.; Ahmadi, A.; Saadati, H.R. Role of PTEN gene in progression of prostate cancer. Urol. J. 2007, 4, 95–100. [Google Scholar]
- Tao, D.L.; Bailey, S.; Beer, T.M.; Foss, E.; Beckett, B.; Fung, A.; Foster, B.R.; Guimaraes, A.; Cetnar, J.P.; Graff, J.N.; et al. Molecular Testing in Patients With Castration-Resistant Prostate Cancer and Its Impact on Clinical Decision Making. JCO Precis. Oncol. 2017, 1–11. [Google Scholar] [CrossRef]
- Geybels, M.S.; Fang, M.; Wright, J.L.; Qu, X.; Bibikova, M.; Klotzle, B.; Fan, J.B.; Feng, Z.; Ostrander, E.A.; Nelson, P.S.; et al. PTEN loss is associated with prostate cancer recurrence and alterations in tumor DNA methylation profiles. Oncotarget 2017, 8, 84338–84348. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, M.; Cunha, I.W.; Coudry, R.A.; Fonseca, F.P.; Torres, C.H.; Soares, F.A.; Squire, J.A. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br. J. Cancer 2007, 97, 678–685. [Google Scholar] [CrossRef]
- Gray, I.C.; Phillips, S.M.; Lee, S.J.; Neoptolemos, J.P.; Weissenbach, J.; Spurr, N.K. Loss of the chromosomal region 10q23-25 in prostate cancer. Cancer Res. 1995, 55, 4800–4803. [Google Scholar]
- Schmitz, M.; Grignard, G.; Margue, C.; Dippel, W.; Capesius, C.; Mossong, J.; Nathan, M.; Giacchi, S.; Scheiden, R.; Kieffer, N. Complete loss of PTEN expression as a possible early prognostic marker for prostate cancer metastasis. Int. J. Cancer 2007, 120, 1284–1292. [Google Scholar] [CrossRef]
- Dreher, T.; Zentgraf, H.; Abel, U.; Kappeler, A.; Michel, M.S.; Bleyl, U.; Grobholz, R. Reduction of PTEN and p27kip1 expression correlates with tumor grade in prostate cancer. Analysis in radical prostatectomy specimens and needle biopsies. Virchows Arch. 2004, 444, 509–517. [Google Scholar] [CrossRef]
- Kazim, Z.; Wahabi, K.; Perwez, A.; Lal, P.; Rizvi, M.A. PTEN Genetic and Epigenetic Alterations Define Distinct Subgroups in North Indian Breast Cancer Patients. Asian Pac. J. Cancer Prev. APJCP 2019, 20, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Liang, F.; Jia, Z.L.; Song, S.T.; Jiang, Z.F. PTEN mutation, methylation and expression in breast cancer patients. Oncol. Lett. 2013, 6, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.M.; Silva, J.M.; Dominguez, G.; Gonzalez, R.; Navarro, A.; Carretero, L.; Provencio, M.; Espana, P.; Bonilla, F. Allelic loss of the PTEN region (10q23) in breast carcinomas of poor pathophenotype. Breast Cancer Res. Treat. 1999, 57, 237–243. [Google Scholar] [CrossRef]
- Tanic, N.; Milovanovic, Z.; Tanic, N.; Dzodic, R.; Juranic, Z.; Susnjar, S.; Plesinac-Karapandzic, V.; Tatic, S.; Dramicanin, T.; Davidovic, R.; et al. The impact of PTEN tumor suppressor gene on acquiring resistance to tamoxifen treatment in breast cancer patients. Cancer Biol. Ther. 2012, 13, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Panigrahi, A.R.; Pinder, S.E.; Chan, S.Y.; Paish, E.C.; Robertson, J.F.; Ellis, I.O. The role of PTEN and its signalling pathways, including AKT, in breast cancer; an assessment of relationships with other prognostic factors and with outcome. J. Pathol. 2004, 204, 93–100. [Google Scholar] [CrossRef]
- Kato, H.; Kato, S.; Kumabe, T.; Sonoda, Y.; Yoshimoto, T.; Kato, S.; Han, S.Y.; Suzuki, T.; Shibata, H.; Kanamaru, R.; et al. Functional evaluation of p53 and PTEN gene mutations in gliomas. Clin. Cancer Res. 2000, 6, 3937–3943. [Google Scholar]
- Wang, S.I.; Puc, J.; Li, J.; Bruce, J.N.; Cairns, P.; Sidransky, D.; Parsons, R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997, 57, 4183–4186. [Google Scholar]
- Abdullah, J.M.; Farizan, A.; Asmarina, K.; Zainuddin, N.; Ghazali, M.M.; Jaafar, H.; Isa, M.N.; Naing, N.N. Association of loss of heterozygosity and PTEN gene abnormalities with paraclinical, clinical modalities and survival time of glioma patients in Malaysia. Asian J. Surg. 2006, 29, 274–282. [Google Scholar] [CrossRef]
- Kurose, K.; Zhou, X.P.; Araki, T.; Cannistra, S.A.; Maher, E.R.; Eng, C. Frequent loss of PTEN expression is linked to elevated phosphorylated Akt levels, but not associated with p27 and cyclin D1 expression, in primary epithelial ovarian carcinomas. Am. J. Pathol. 2001, 158, 2097–2106. [Google Scholar] [CrossRef]
- Obata, K.; Morland, S.J.; Watson, R.H.; Hitchcock, A.; Chenevix-Trench, G.; Thomas, E.J.; Campbell, I.G. Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res. 1998, 58, 2095–2097. [Google Scholar] [PubMed]
- Saito, M.; Okamoto, A.; Kohno, T.; Takakura, S.; Shinozaki, H.; Isonishi, S.; Yasuhara, T.; Yoshimura, T.; Ohtake, Y.; Ochiai, K.; et al. Allelic imbalance and mutations of the PTEN gene in ovarian cancer. Int. J. Cancer 2000, 85, 160–165. [Google Scholar] [CrossRef]
- Abubaker, J.; Bavi, P.; Al-Haqawi, W.; Jehan, Z.; Munkarah, A.; Uddin, S.; Al-Kuraya, K.S. PIK3CA alterations in Middle Eastern ovarian cancers. Mol. Cancer 2009, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.C.; Santiago, I.; Trinh, A.; Xian, J.; Guo, A.; Sayal, K.; Jimenez-Linan, M.; Deen, S.; Driver, K.; Mack, M.; et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome Biol. 2014, 15, 526. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, N.; Nagai, H.; Bando, K.; Koyama, M.; Matsumoto, S.; Tajiri, T.; Onda, M.; Fujimoto, J.; Ueki, T.; Konishi, N.; et al. PTEN/MMAC1 mutations in hepatocellular carcinomas: Somatic inactivation of both alleles in tumors. Jpn. J. Cancer Res. 1999, 90, 413–418. [Google Scholar] [CrossRef]
- Rahman, M.A.; Kyriazanos, I.D.; Ono, T.; Yamanoi, A.; Kohno, H.; Tsuchiya, M.; Nagasue, N. Impact of PTEN expression on the outcome of hepatitis C virus-positive cirrhotic hepatocellular carcinoma patients: Possible relationship with COX II and inducible nitric oxide synthase. Int. J. Cancer 2002, 100, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.H.; Cong, W.M.; Xian, Z.H.; Wu, M.C. Clinicopathological significance of loss of heterozygosity and microsatellite instability in hepatocellular carcinoma in China. World J. Gastroenterol. 2005, 11, 3034–3039. [Google Scholar] [CrossRef]
- Dong-Dong, L.; Xi-Ran, Z.; Xiang-Rong, C. Expression and significance of new tumor suppressor gene PTEN in primary liver cancer. J. Cell. Mol. Med. 2003, 7, 67–71. [Google Scholar] [CrossRef]
- Marsit, C.J.; Zheng, S.; Aldape, K.; Hinds, P.W.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. PTEN expression in non-small-cell lung cancer: Evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum. Pathol. 2005, 36, 768–776. [Google Scholar] [CrossRef]
- Yoo, S.B.; Xu, X.; Lee, H.J.; Jheon, S.; Lee, C.T.; Choe, G.; Chung, J.H. Loss of PTEN Expression is an Independent Poor Prognostic Factor in Non-small Cell Lung Cancer. J. Pathol. Transl. Med. 2011, 45, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Yanagawa, N.; Leduc, C.; Kohler, D.; Saieg, M.A.; John, T.; Sykes, J.; Yoshimoto, M.; Pintilie, M.; Squire, J.; Shepherd, F.A.; et al. Loss of phosphatase and tensin homolog protein expression is an independent poor prognostic marker in lung adenocarcinoma. J. Thorac. Oncol. 2012, 7, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Kim, M.J.; Jeon, H.S.; Choi, J.E.; Kim, D.S.; Lee, E.B.; Cha, S.I.; Yoon, G.S.; Kim, C.H.; Jung, T.H.; et al. PTEN mutations and relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small cell lung cancers. Lung Cancer 2010, 69, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Atreya, C.E.; Sangale, Z.; Xu, N.; Matli, M.R.; Tikishvili, E.; Welbourn, W.; Stone, S.; Shokat, K.M.; Warren, R.S. PTEN expression is consistent in colorectal cancer primaries and metastases and associates with patient survival. Cancer Med. 2013, 2, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Dicuonzo, G.; Angeletti, S.; Garcia-Foncillas, J.; Brugarolas, A.; Okrouzhnov, Y.; Santini, D.; Tonini, G.; Lorino, G.; De Cesaris, M.; Baldi, A. Colorectal carcinomas and PTEN/MMAC1 gene mutations. Clin. Cancer Res. 2001, 7, 4049–4053. [Google Scholar] [PubMed]
- Nassif, N.T.; Lobo, G.P.; Wu, X.; Henderson, C.J.; Morrison, C.D.; Eng, C.; Jalaludin, B.; Segelov, E. PTEN mutations are common in sporadic microsatellite stable colorectal cancer. Oncogene 2004, 23, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negri, F.V.; Bozzetti, C.; Lagrasta, C.A.; Crafa, P.; Bonasoni, M.P.; Camisa, R.; Pedrazzi, G.; Ardizzoni, A. PTEN status in advanced colorectal cancer treated with cetuximab. Br. J. Cancer 2010, 102, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Aggerholm, A.; Gronbaek, K.; Guldberg, P.; Hokland, P. Mutational analysis of the tumour suppressor gene MMAC1/PTEN in malignant myeloid disorders. Eur. J. Haematol. 2000, 65, 109–113. [Google Scholar] [CrossRef]
- Liu, T.C.; Lin, P.M.; Chang, J.G.; Lee, J.P.; Chen, T.P.; Lin, S.F. Mutation analysis of PTEN/MMAC1 in acute myeloid leukemia. Am. J. Hematol. 2000, 63, 170–175. [Google Scholar] [CrossRef]
- Mendes, R.D.; Sarmento, L.M.; Cante-Barrett, K.; Zuurbier, L.; Buijs-Gladdines, J.G.; Povoa, V.; Smits, W.K.; Abecasis, M.; Yunes, J.A.; Sonneveld, E.; et al. PTEN microdeletions in T-cell acute lymphoblastic leukemia are caused by illegitimate RAG-mediated recombination events. Blood 2014, 124, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Jenkinson, S.; Kirkwood, A.A.; Goulden, N.; Vora, A.; Linch, D.C.; Gale, R.E. Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia 2016, 30, 39–47. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson Gedman, A.; Chen, Q.; Kugel Desmoulin, S.; Ge, Y.; LaFiura, K.; Haska, C.L.; Cherian, C.; Devidas, M.; Linda, S.B.; Taub, J.W.; et al. The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-cell acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Leukemia 2009, 23, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.; Gatalica, Z.; Knezetic, J.; Reddy, S.; Nathan, C.A.; Javadi, N.; Teknos, T. Molecular profiling of head and neck squamous cell carcinoma. Head Neck 2016, 38 (Suppl. 1), E1625–E1638. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Guthrie, V.B.; Masica, D.L.; Tokheim, C.; Kang, H.; Richmon, J.; Agrawal, N.; Fakhry, C.; Quon, H.; Subramaniam, R.M.; et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann. Oncol. 2015, 26, 1216–1223. [Google Scholar] [CrossRef]
- Squarize, C.H.; Castilho, R.M.; Abrahao, A.C.; Molinolo, A.; Lingen, M.W.; Gutkind, J.S. PTEN deficiency contributes to the development and progression of head and neck cancer. Neoplasia 2013, 15, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Snietura, M.; Jaworska, M.; Mlynarczyk-Liszka, J.; Goraj-Zajac, A.; Piglowski, W.; Lange, D.; Wozniak, G.; Nowara, E.; Suwinski, R. PTEN as a prognostic and predictive marker in postoperative radiotherapy for squamous cell cancer of the head and neck. PLoS ONE 2012, 7, e33396. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, M.; Lorenz, G.; Kleist, B. Detection of new PTEN/MMAC1 mutations in head and neck squamous cell carcinomas with loss of chromosome 10. Cancer Genet. Cytogenet. 2002, 132, 20–24. [Google Scholar] [CrossRef]
- Okami, K.; Wu, L.; Riggins, G.; Cairns, P.; Goggins, M.; Evron, E.; Halachmi, N.; Ahrendt, S.A.; Reed, A.L.; Hilgers, W.; et al. Analysis of PTEN/MMAC1 alterations in aerodigestive tract tumors. Cancer Res. 1998, 58, 509–511. [Google Scholar] [PubMed]
- Fuchs, E.; Chen, T. A matter of life and death: Self-renewal in stem cells. EMBO Rep. 2013, 14, 39–48. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer stem cells and self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Wicha, M.S. Selective targeting of cancer stem cells: A new concept in cancer therapeutics. BioDrugs 2007, 21, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagi, S.; Kishimoto, H.; Kawahara, K.; Sasaki, T.; Sasaki, M.; Nishio, M.; Yajima, N.; Hamada, K.; Horie, Y.; Kubo, H.; et al. Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J. Clin. Investig. 2007, 117, 2929–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, S.; Yuan, G.; Liu, X.; Ren, R.; Li, J.; Zhang, W.; Wu, J.; Xu, X.; Fu, L.; Li, Y.; et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat. Commun. 2015, 6, 10068. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Xin, L.; Morim, A.; Lawson, D.; Witte, O.; Wu, H. Lin-Sca-1+CD49fhigh stem/progenitors are tumor-initiating cells in the Pten-null prostate cancer model. Cancer Res. 2009, 69, 8555–8562. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Kim, S.; Salamone, R.J.; Walker, J.R.; Maira, S.M.; Garcia-Echeverria, C.; Schultz, P.G.; Reddy, V.A. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc. Natl. Acad. Sci. USA 2009, 106, 268–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkaya, H.; Paulson, A.; Charafe-Jauffret, E.; Ginestier, C.; Brown, M.; Dutcher, J.; Clouthier, S.G.; Wicha, M.S. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009, 7, e1000121. [Google Scholar] [CrossRef]
- Chen, R.; Nishimura, M.C.; Bumbaca, S.M.; Kharbanda, S.; Forrest, W.F.; Kasman, I.M.; Greve, J.M.; Soriano, R.H.; Gilmour, L.L.; Rivers, C.S.; et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar] [CrossRef]
- Malanga, D.; De Marco, C.; Guerriero, I.; Colelli, F.; Rinaldo, N.; Scrima, M.; Mirante, T.; De Vitis, C.; Zoppoli, P.; Ceccarelli, M.; et al. The Akt1/IL-6/STAT3 pathway regulates growth of lung tumor initiating cells. Oncotarget 2015, 6, 42667–42686. [Google Scholar] [CrossRef]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef]
- Abou-Kheir, W.G.; Hynes, P.G.; Martin, P.L.; Pierce, R.; Kelly, K. Characterizing the contribution of stem/progenitor cells to tumorigenesis in the Pten-/-TP53-/- prostate cancer model. Stem Cells 2010, 28, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Abou-Kheir, W.; Hynes, P.G.; Martin, P.; Yin, J.J.; Liu, Y.N.; Seng, V.; Lake, R.; Spurrier, J.; Kelly, K. Self-renewing Pten-/- TP53-/- protospheres produce metastatic adenocarcinoma cell lines with multipotent progenitor activity. PLoS ONE 2011, 6, e26112. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Yang, G.D.; Huang, T.J.; Li, R.; Chu, Q.Q.; Xu, L.; Wang, M.S.; Cai, M.D.; Zhong, L.; Wei, H.J.; et al. EB-virus latent membrane protein 1 potentiates the stemness of nasopharyngeal carcinoma via preferential activation of PI3K/AKT pathway by a positive feedback loop. Oncogene 2016, 35, 3419–3431. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.C.; Jiao, M.; Wu, T.; Jing, L.; Cui, J.; Guo, H.; Tian, T.; Ruan, Z.P.; Wei, Y.C.; Jiang, L.L.; et al. Polycomb complex protein BMI-1 promotes invasion and metastasis of pancreatic cancer stem cells by activating PI3K/AKT signaling, an ex vivo, in vitro, and in vivo study. Oncotarget 2016, 7, 9586–9599. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.J.; Rho, J.K.; Kim, Y.M.; Jung, J.E.; Jin, Y.B.; Ko, Y.G.; Lee, J.S.; Lee, S.J.; Lee, J.C.; Park, M.J. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene 2013, 32, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ding, J.; Chen, C.; Sun, W.; Ning, B.F.; Wen, W.; Huang, L.; Han, T.; Yang, W.; Wang, C.; et al. Hepatic transforming growth factor beta gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 2012, 56, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Al-Dhfyan, A.; Alhoshani, A.; Korashy, H.M. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and beta-Catenin and Akt activation. Mol. Cancer 2017, 16, 14. [Google Scholar] [CrossRef]
- Salah, M.; Nishimoto, Y.; Kohno, S.; Kondoh, A.; Kitajima, S.; Muranaka, H.; Nishiuchi, T.; Ibrahim, A.; Yoshida, A.; Takahashi, C. An in vitro system to characterize prostate cancer progression identified signaling required for self-renewal. Mol. Carcinog. 2016, 55, 1974–1989. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, Y.; Liu, Y.; Zhou, M.; Lu, Y.; Yuan, L.; Zhang, C.; Hong, M.; Wang, S.; Li, X. MiR-106b induces cell radioresistance via the PTEN/PI3K/AKT pathways and p21 in colorectal cancer. J. Transl. Med. 2015, 13, 252. [Google Scholar] [CrossRef]
- Bahena-Ocampo, I.; Espinosa, M.; Ceballos-Cancino, G.; Lizarraga, F.; Campos-Arroyo, D.; Schwarz, A.; Garcia-Lopez, P.; Maldonado, V.; Melendez-Zajgla, J. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep. 2016, 17, 648–658. [Google Scholar] [CrossRef]
- Banasavadi-Siddegowda, Y.K.; Russell, L.; Frair, E.; Karkhanis, V.A.; Relation, T.; Yoo, J.Y.; Zhang, J.; Sif, S.; Imitola, J.; Baiocchi, R.; et al. PRMT5-PTEN molecular pathway regulates senescence and self-renewal of primary glioblastoma neurosphere cells. Oncogene 2017, 36, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Chen, Y.; Li, D.; Li, S. Role of Pten in leukemia stem cells. Oncotarget 2010, 1, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Cardenas, A.; Chen, H.; Garcia, C.; Guo, W.; Bradner, J.; Wu, H. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res. 2014, 74, 7048–7059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Setoguchi, T.; Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. USA 2004, 101, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Zhou, J.; Claypool, K.; Tang, D.G. Side Population Is Enriched in Tumorigenic, Stem-Like Cancer Cells, whereas ABCG2+ and ABCG2− Cancer Cells Are Similarly Tumorigenic. Cancer Res. 2005, 65, 6207–6219. [Google Scholar] [CrossRef]
- Qiang, L.; Yang, Y.; Ma, Y.J.; Chen, F.H.; Zhang, L.B.; Liu, W.; Qi, Q.; Lu, N.; Tao, L.; Wang, X.T.; et al. Isolation and characterization of cancer stem like cells in human glioblastoma cell lines. Cancer Lett. 2009, 279, 13–21. [Google Scholar] [CrossRef]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, B.B. Stress response of glioblastoma cells mediated by miR-17-5p targeting PTEN and the passenger strand miR-17-3p targeting MDM2. Oncotarget 2012, 3, 1653–1668. [Google Scholar] [CrossRef]
- Lo, J.F.; Yu, C.C.; Chiou, S.H.; Huang, C.Y.; Jan, C.I.; Lin, S.C.; Liu, C.J.; Hu, W.Y.; Yu, Y.H. The epithelial-mesenchymal transition mediator S100A4 maintains cancer-initiating cells in head and neck cancers. Cancer Res. 2011, 71, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Dong, Z.; Chen, Y.; Yang, L.; Lai, D. Enrichment of ovarian cancer stem-like cells is associated with epithelial to mesenchymal transition through an miRNA-activated AKT pathway. Cell Prolif. 2013, 46, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lu, Y.; Wang, H.; Han, X.; Mao, J.; Li, J.; Yu, L.; Wang, B.; Fan, S.; Yu, X.; et al. miR-221/222 enhance the tumorigenicity of human breast cancer stem cells via modulation of PTEN/Akt pathway. Biomed. Pharmacother. 2016, 79, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Li, C.; Wang, S.; Li, Y.; Wen, B.; Zhang, Y.; Liang, K.; Yao, J.; Ye, Y.; Hsiao, H.; et al. LncRNAs-directed PTEN enzymatic switch governs epithelial-mesenchymal transition. Cell Res. 2019, 29, 286–304. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, M.V.; Dahiya, S.; Danielson, L.S.; Segura, M.F.; Vales-Lara, F.M.; Menendez, S.; Popiolek, D.; Mittal, K.; Wei, J.J.; Zavadil, J.; et al. Dual Pten/Tp53 suppression promotes sarcoma progression by activating Notch signaling. Am. J. Pathol. 2013, 182, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.P.; Lin, T.P.; Li, P.C.; Geary, L.A.; Chen, K.; Vaikari, V.P.; Wu, J.B.; Lin, C.H.; Gross, M.E.; Shih, J.C. Loss of MAOA in epithelia inhibits adenocarcinoma development, cell proliferation and cancer stem cells in prostate. Oncogene 2018, 37, 5175–5190. [Google Scholar] [CrossRef]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef]
- Peng, C.; Chen, Y.; Yang, Z.; Zhang, H.; Osterby, L.; Rosmarin, A.G.; Li, S. PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood 2010, 115, 626–635. [Google Scholar] [CrossRef]
- Guo, W.; Schubbert, S.; Chen, J.Y.; Valamehr, B.; Mosessian, S.; Shi, H.; Dang, N.H.; Garcia, C.; Theodoro, M.F.; Varella-Garcia, M.; et al. Suppression of leukemia development caused by PTEN loss. Proc. Natl. Acad. Sci. USA 2011, 108, 1409–1414. [Google Scholar] [CrossRef] [Green Version]
- Yue, F.; Bi, P.; Wang, C.; Shan, T.; Nie, Y.; Ratliff, T.L.; Gavin, T.P.; Kuang, S. Pten is necessary for the quiescence and maintenance of adult muscle stem cells. Nat. Commun. 2017, 8, 14328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groszer, M.; Erickson, R.; Scripture-Adams, D.D.; Lesche, R.; Trumpp, A.; Zack, J.A.; Kornblum, H.I.; Liu, X.; Wu, H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 2001, 294, 2186–2189. [Google Scholar] [CrossRef] [PubMed]
- Groszer, M.; Erickson, R.; Scripture-Adams, D.D.; Dougherty, J.D.; Le Belle, J.; Zack, J.A.; Geschwind, D.H.; Liu, X.; Kornblum, H.I.; Wu, H. PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry. Proc. Natl. Acad. Sci. USA 2006, 103, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.G.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Investig. 2010, 120, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Hwang, H.J.; Kang, D.; Park, H.A.; Lee, H.C.; Jeong, D.; Lee, K.; Park, H.J.; Ko, Y.G.; Lee, J.S. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 2019, 38, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Hall, B.; Sun, Z.J.; Molinolo, A.; Chen, W.; Gutkind, J.S.; Waes, C.V.; Kulkarni, A.B. Loss of TGF-beta signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012, 31, 3322–3332. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Kohnoh, T.; Hashimoto, N.; Ando, A.; Sakamoto, K.; Miyazaki, S.; Aoyama, D.; Kusunose, M.; Kimura, M.; Omote, N.; Imaizumi, K.; et al. Hypoxia-induced modulation of PTEN activity and EMT phenotypes in lung cancers. Cancer Cell Int. 2016, 16, 33. [Google Scholar] [CrossRef]
- Aoyama, D.; Hashimoto, N.; Sakamoto, K.; Kohnoh, T.; Kusunose, M.; Kimura, M.; Ogata, R.; Imaizumi, K.; Kawabe, T.; Hasegawa, Y. Involvement of TGFbeta-induced phosphorylation of the PTEN C-terminus on TGFbeta-induced acquisition of malignant phenotypes in lung cancer cells. PLoS ONE 2013, 8, e81133. [Google Scholar] [CrossRef]
- Wu, J.B.; Shao, C.; Li, X.; Li, Q.; Hu, P.; Shi, C.; Li, Y.; Chen, Y.T.; Yin, F.; Liao, C.P.; et al. Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J. Clin. Investig. 2014, 124, 2891–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaraiz-Rodriguez, M.; Tabernero, M.D.; Gonzalez-Tablas, M.; Otero, A.; Orfao, A.; Medina, J.M.; Tabernero, A. A Short Region of Connexin43 Reduces Human Glioma Stem Cell Migration, Invasion, and Survival through Src, PTEN, and FAK. Stem Cell Rep. 2017, 9, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.H.; Hao, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Emerging roles of radioresistance in prostate cancer metastasis and radiation therapy. Cancer Metastasis Rev. 2014, 33, 469–496. [Google Scholar] [CrossRef] [PubMed]
- Untch, M.; Fasching, P.A.; Konecny, G.E.; Hasmuller, S.; Lebeau, A.; Kreienberg, R.; Camara, O.; Muller, V.; du Bois, A.; Kuhn, T.; et al. Pathologic complete response after neoadjuvant chemotherapy plus trastuzumab predicts favorable survival in human epidermal growth factor receptor 2-overexpressing breast cancer: Results from the TECHNO trial of the AGO and GBG study groups. J. Clin. Oncol. 2011, 29, 3351–3357. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A. Compensatory pathways in oncogenic kinase signaling and resistance to targeted therapies: Six degrees of separation. Cancer Discov. 2012, 2, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Wyatt, D.; Bocchetta, M.; Li, J.; Filipovic, A.; Green, A.; Peiffer, D.S.; Fuqua, S.; Miele, L.; Albain, K.S.; et al. Notch-1-PTEN-ERK1/2 signaling axis promotes HER2+ breast cancer cell proliferation and stem cell survival. Oncogene 2018, 37, 4489–4504. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Esteva, F.J. Trastuzumab: Triumphs and tribulations. Oncogene 2007, 26, 3637–3643. [Google Scholar] [CrossRef]
- Burnett, J.P.; Korkaya, H.; Ouzounova, M.D.; Jiang, H.; Conley, S.J.; Newman, B.W.; Sun, L.; Connarn, J.N.; Chen, C.S.; Zhang, N.; et al. Trastuzumab resistance induces EMT to transform HER2(+) PTEN(-) to a triple negative breast cancer that requires unique treatment options. Sci. Rep. 2015, 5, 15821. [Google Scholar] [CrossRef]
- Sun, L.; Burnett, J.; Gasparyan, M.; Xu, F.; Jiang, H.; Lin, C.C.; Myers, I.; Korkaya, H.; Liu, Y.; Connarn, J.; et al. Novel cancer stem cell targets during epithelial to mesenchymal transition in PTEN-deficient trastuzumab-resistant breast cancer. Oncotarget 2016, 7, 51408–51422. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Chinratanalab, W.; Ritter, C.A.; King, W.; Seelig, S.; Arteaga, C.L. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002, 62, 4132–4141. [Google Scholar]
- Ebbesen, S.H.; Scaltriti, M.; Bialucha, C.U.; Morse, N.; Kastenhuber, E.R.; Wen, H.Y.; Dow, L.E.; Baselga, J.; Lowe, S.W. Pten loss promotes MAPK pathway dependency in HER2/neu breast carcinomas. Proc. Natl. Acad. Sci. USA 2016, 113, 3030–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Wu, H. PTEN, stem cells, and cancer stem cells. J. Biol. Chem. 2009, 284, 11755–11759. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.S.; Gilliland, D.G.; Morrison, S.J. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 2010, 7, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Grimaldi, C.; Ricci, F.; Tazzari, P.L.; Evangelisti, C.; Ognibene, A.; Battistelli, M.; Falcieri, E.; Melchionda, F.; Pession, A.; et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. 2010, 70, 8097–8107. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, L.; Wu, Y.; Dong, B.; Guo, W.; Wang, M.; Yang, L.; Fan, X.; Tang, Y.; Liu, N.; et al. T-ALL leukemia stem cell ‘stemness’ is epigenetically controlled by the master regulator SPI1. eLife 2018, 7, e38314. [Google Scholar] [CrossRef]
- Antony-Debre, I.; Paul, A.; Leite, J.; Mitchell, K.; Kim, H.M.; Carvajal, L.A.; Todorova, T.I.; Huang, K.; Kumar, A.; Farahat, A.A.; et al. Pharmacological inhibition of the transcription factor PU.1 in leukemia. J. Clin. Investig. 2017, 127, 4297–4313. [Google Scholar] [CrossRef] [Green Version]
- Munde, M.; Wang, S.; Kumar, A.; Stephens, C.E.; Farahat, A.A.; Boykin, D.W.; Wilson, W.D.; Poon, G.M. Structure-dependent inhibition of the ETS-family transcription factor PU.1 by novel heterocyclic diamidines. Nucleic Acids Res. 2014, 42, 1379–1390. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular carcinoma: An epidemiologic view. J. Clin. Gastroenterol. 2002, 35, S72–S78. [Google Scholar] [CrossRef]
- Cervello, M.; McCubrey, J.A.; Cusimano, A.; Lampiasi, N.; Azzolina, A.; Montalto, G. Targeted therapy for hepatocellular carcinoma: Novel agents on the horizon. Oncotarget 2012, 3, 236–260. [Google Scholar] [CrossRef] [PubMed]
- Kern, M.A.; Schubert, D.; Sahi, D.; Schoneweiss, M.M.; Moll, I.; Haugg, A.M.; Dienes, H.P.; Breuhahn, K.; Schirmacher, P. Proapoptotic and antiproliferative potential of selective cyclooxygenase-2 inhibitors in human liver tumor cells. Hepatology 2002, 36, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Leng, J.; Han, C.; Demetris, A.J.; Michalopoulos, G.K.; Wu, T. Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: Evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology 2003, 38, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Ding, W.; Dang, H.; Jiang, Y.; Rountree, C.B. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 2011, 54, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, T.H.; Chan, H.H.; Kuo, H.M.; Liu, L.F.; Hu, T.H.; Sun, C.K.; Kung, M.L.; Lin, S.W.; Wang, E.M.; Ma, Y.L.; et al. Celecoxib suppresses hepatoma stemness and progression by up-regulating PTEN. Oncotarget 2014, 5, 1475–1490. [Google Scholar] [CrossRef] [PubMed]
- Rountree, C.B.; Ding, W.; He, L.; Stiles, B. Expansion of CD133-expressing liver cancer stem cells in liver-specific phosphatase and tensin homolog deleted on chromosome 10-deleted mice. Stem Cells 2009, 27, 290–299. [Google Scholar] [CrossRef]
- Tang, Y.; Kitisin, K.; Jogunoori, W.; Li, C.; Deng, C.X.; Mueller, S.C.; Ressom, H.W.; Rashid, A.; He, A.R.; Mendelson, J.S.; et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 2445–2450. [Google Scholar] [CrossRef]
- Lee, T.K.; Castilho, A.; Cheung, V.C.; Tang, K.H.; Ma, S.; Ng, I.O. Lupeol targets liver tumor-initiating cells through phosphatase and tensin homolog modulation. Hepatology 2011, 53, 160–170. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Giaume, C.; Koulakoff, A.; Roux, L.; Holcman, D.; Rouach, N. Astroglial networks: A step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci. 2010, 11, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sanchez, A.; Jaraiz-Rodriguez, M.; Dominguez-Prieto, M.; Herrero-Gonzalez, S.; Medina, J.M.; Tabernero, A. Connexin43 recruits PTEN and Csk to inhibit c-Src activity in glioma cells and astrocytes. Oncotarget 2016, 7, 49819–49833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellino, R.C.; Barwick, B.G.; Schniederjan, M.; Buss, M.C.; Becher, O.; Hambardzumyan, D.; Macdonald, T.J.; Brat, D.J.; Durden, D.L. Heterozygosity for Pten promotes tumorigenesis in a mouse model of medulloblastoma. PLoS ONE 2010, 5, e10849. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.R.; Joshi, S.; Zulcic, M.; Alcaraz, M.; Garlich, J.R.; Morales, G.A.; Cho, Y.J.; Bao, L.; Levy, M.L.; Newbury, R.; et al. PI-3K Inhibitors Preferentially Target CD15+ Cancer Stem Cell Population in SHH Driven Medulloblastoma. PLoS ONE 2016, 11, e0150836. [Google Scholar] [CrossRef] [PubMed]
- Grubb, R.L., 3rd; Kibel, A.S. Prostate cancer: Screening, diagnosis and management in 2007. Mo. Med. 2007, 104, 408–413. [Google Scholar] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Elliott, J.; Salamone, R.J.; Kim, S.; Aimone, L.J.; Walker, J.R.; Watson, J.; Sauveur-Michel, M.; Garcia-Echeverria, C.; Cho, C.Y.; et al. Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clin. Cancer Res. 2010, 16, 5692–5702. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364, eaau0159. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Matteoni, S.; Sacconi, A.; De Luca, T.; Bazzichetto, C.; Corbo, V.; Simbolo, M.; Sperduti, I.; et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci. Rep. 2017, 7, 43013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, Y.; Scopelliti, A.; Cammareri, P.; Todaro, M.; Iovino, F.; Ricci-Vitiani, L.; Gulotta, G.; Dieli, F.; de Maria, R.; Stassi, G. Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology 2011, 140, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Mollinari, C.; di Martino, S.; Biffoni, M.; Pilozzi, E.; Pagliuca, A.; de Stefano, M.C.; Circo, R.; Merlo, D.; De Maria, R.; et al. Thymosin beta4 targeting impairs tumorigenic activity of colon cancer stem cells. FASEB J. 2010, 24, 4291–4301. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.K.; Choi, Y.J.; Lee, J.K.; Ryoo, B.Y.; Na, I.I.; Yang, S.H.; Kim, C.H.; Lee, J.C. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer 2009, 63, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Faversani, A.; Amatori, S.; Augello, C.; Colombo, F.; Porretti, L.; Fanelli, M.; Ferrero, S.; Palleschi, A.; Pelicci, P.G.; Belloni, E.; et al. miR-494-3p is a novel tumor driver of lung carcinogenesis. Oncotarget 2017, 8, 7231–7247. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Zhou, X.; Li, S.; Qin, Y.; Chen, Y.; Liu, H. Inhibition of miR-23a increases the sensitivity of lung cancer stem cells to erlotinib through PTEN/PI3K/Akt pathway. Oncol. Rep. 2017, 38, 3064–3070. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Site | Malignancy Type | Molecular Mechanism(s) of PTEN Alteration and Incidence (%) | Reference |
|---|---|---|---|
| Prostate | Prostate cancer | Mutation: 12–26% LOH: 10–62% Reduced expression: 27–95% | [75,76,77,78,79,80,81] |
| Breast | Breast cancer | Mutation: <7% LOH: 29–63% Reduced expression: 8–55% | [82,83,84,85,86] |
| Brain | Glioma | Mutation: 12–44% LOH: 32–84% Reduced expression: 69% | [42,87,88,89] |
| Ovary | Ovarian carcinoma | Mutation: <9% LOH: 32–61% Reduced expression: 23–55% | [90,91,92,93,94] |
| Liver | Liver cancer | Mutation: <5% LOH: 27–79% Reduced expression: 30–63% | [95,96,97,98] |
| Lung | Non-small-cell lung cancer | Mutation: <5% LOH: 3–19% Reduced expression: 41–73% | [99,100,101,102] |
| Colorectum | Colorectal cancer | Mutation: 17–20% LOH: 20–30% Reduced expression: 12% | [103,104,105,106] |
| Blood | Myeloid leukemia | Deletion: rare LOH: Absent Reduced expression: 24% | [107,108] |
| Lymphoid leukemia | Deletion: 8–63% LOH: NA Reduced expression: 6–17% | [109,110,111,112] | |
| Head-neck | Head and neck squamous cell carcinoma | Deletion: 2–23% LOH: 41% Reduced expression: 31–60% | [113,114,115,116,117,118] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luongo, F.; Colonna, F.; Calapà, F.; Vitale, S.; Fiori, M.E.; De Maria, R. PTEN Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers 2019, 11, 1076. https://doi.org/10.3390/cancers11081076
Luongo F, Colonna F, Calapà F, Vitale S, Fiori ME, De Maria R. PTEN Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers. 2019; 11(8):1076. https://doi.org/10.3390/cancers11081076
Chicago/Turabian StyleLuongo, Francesca, Francesca Colonna, Federica Calapà, Sara Vitale, Micol E. Fiori, and Ruggero De Maria. 2019. "PTEN Tumor-Suppressor: The Dam of Stemness in Cancer" Cancers 11, no. 8: 1076. https://doi.org/10.3390/cancers11081076
APA StyleLuongo, F., Colonna, F., Calapà, F., Vitale, S., Fiori, M. E., & De Maria, R. (2019). PTEN Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers, 11(8), 1076. https://doi.org/10.3390/cancers11081076