PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around

 , , , , , , , ,
, , , , , , , ,  ,
,  and
and
Abstract
:1. Introduction
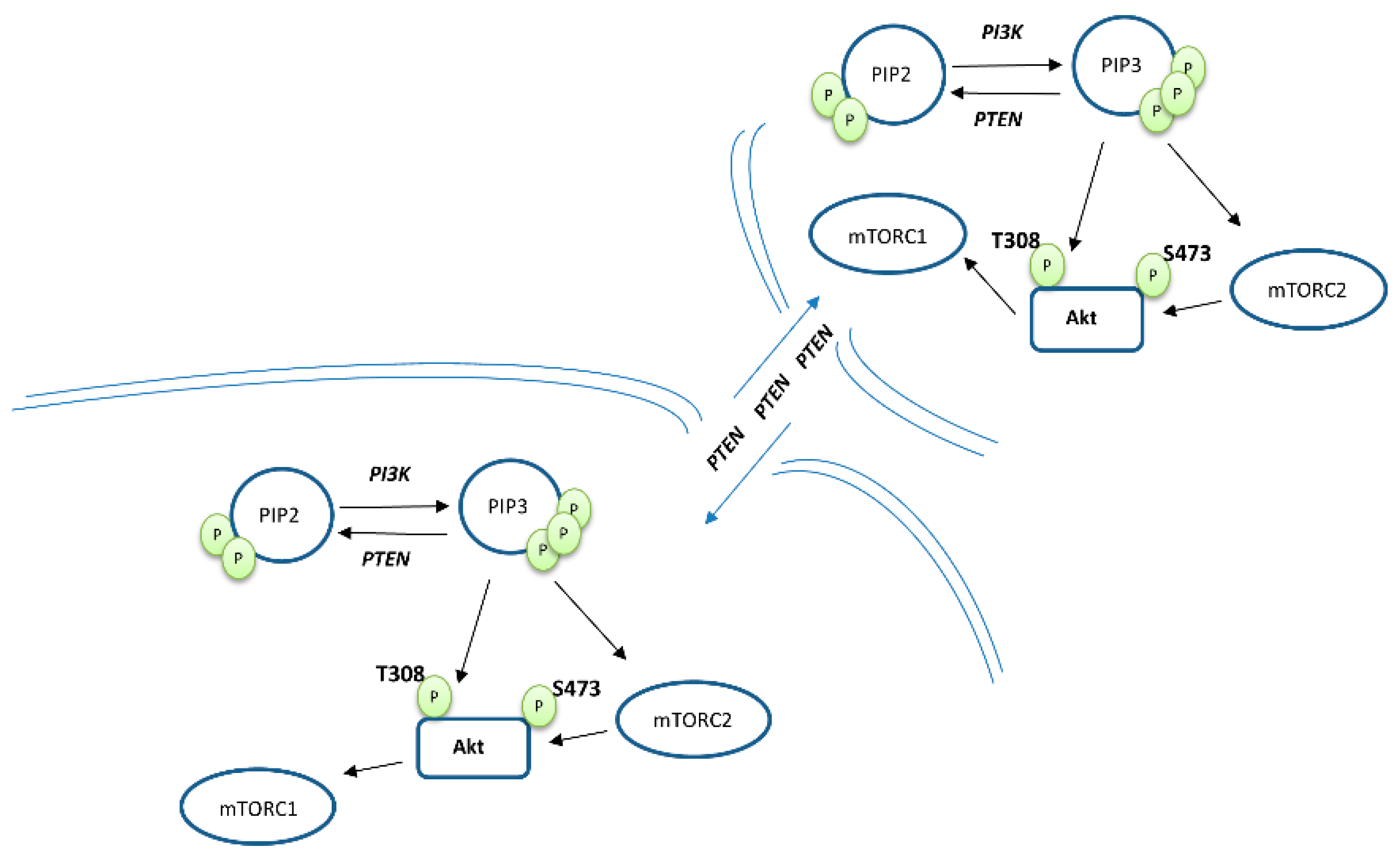
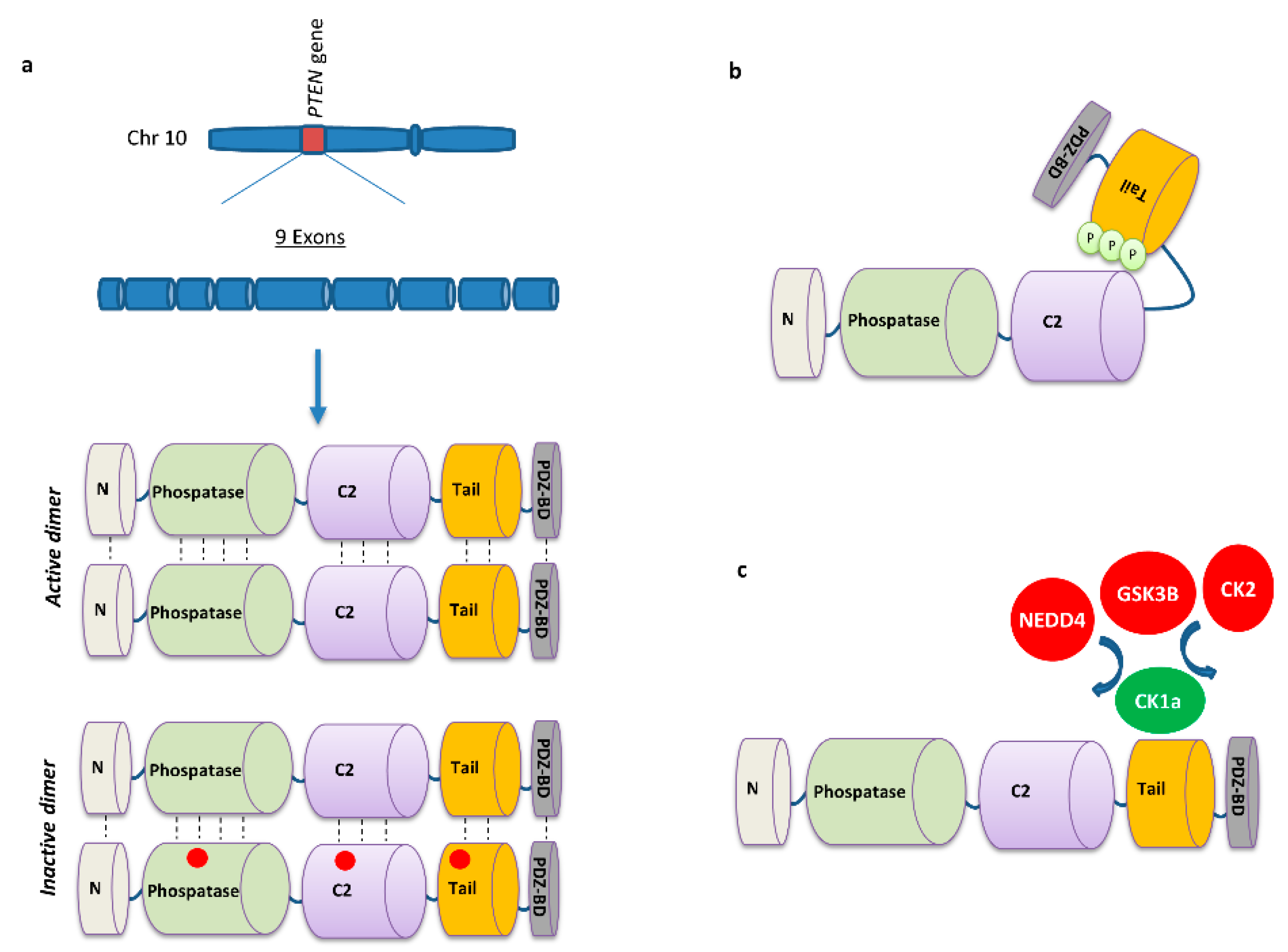
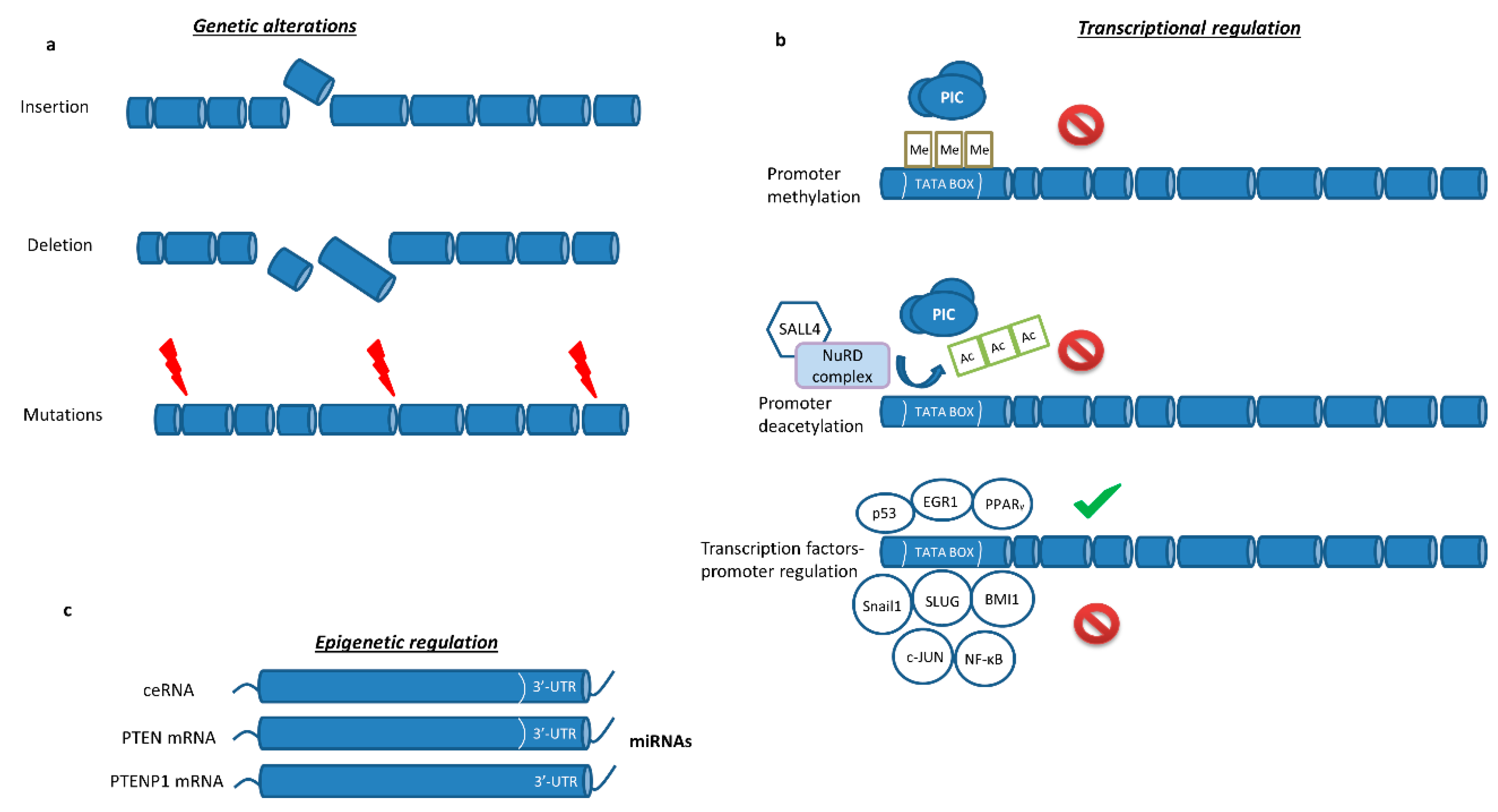
2. PTEN Biology
2.1. PTEN Epigenetic Regulation
2.2. PTEN Post-Transcriptional Modulation by Non-Coding RNAs
2.3. PTEN Post-Translational Regulation
2.4. PTEN-Transcription Factors Interaction
3. PTEN Dysregulation Initiates Oncogenesis
4. PTEN as an Inhibitory Factor for Metastasis
5. PTEN Status and Clinical Implications in Lung Cancer
5.1. PTEN Genetic Status in Lung Cancer
5.2. PTEN Protein Status in Lung Cancer
5.3. PI3K/mTOR/Akt Inhibition in Lung Cancer
6. PTEN-Mediated Resistance to Targeted Therapy
7. PTEN Role in Tumor Microenvironment and Immunotherapy Sensitivity
8. Conclusions
Funding
Conflicts of Interest
References
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Karachaliou, N.; Morales-Espinosa, D.; Costa, C.; Molina, M.A.; Sansano, I.; Gasco, A.; Viteri, S.; Massuti, B.; Wei, J.; et al. Adaptive resistance to targeted therapies in cancer. Transl. Lung Cancer Res. 2013, 2, 152–159. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Alimonti, A.; Carracedo, A.; Clohessy, J.G.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Shen-Li, H.; Koujak, S.; Szablocs, M.; Parsons, R. Reduction of Pten dose leads to neoplastic development in multiple organs of Pten (shRNA) mice. Cancer Biol. Ther. 2010, 10, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef]
- Gkountakos, A.; Pilotto, S.; Mafficini, A.; Vicentini, C.; Simbolo, M.; Milella, M.; Tortora, G.; Scarpa, A.; Bria, E.; Corbo, V. Unmasking the impact of Rictor in cancer: Novel insights of mTORC2 complex. Carcinogenesis 2018, 39, 971–980. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef]
- Rahdar, M.; Inoue, T.; Meyer, T.; Zhang, J.; Vazquez, F.; Devreotes, P.N. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc. Natl. Acad. Sci. USA 2009, 106, 480–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putz, U.; Howitt, J.; Doan, A.; Goh, C.P.; Low, L.H.; Silke, J.; Tan, S.S. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal. 2012, 5, ra70. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 2013, 341, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Lavictoire, S.J.; Gont, A.; Julian, L.M.; Stanford, W.L.; Vlasschaert, C.; Gray, D.A.; Jomaa, D.; Lorimer, I.A.J. Engineering PTEN-L for Cell-Mediated Delivery. Mol. Ther. Methods Clin. Dev. 2018, 9, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R.; Baker, S.J.; Barata, J.T.; Carracedo, A.; Cid, V.J.; Chin-Sang, I.D.; Dave, V.; den Hertog, J.; Devreotes, P.; Eickholt, B.J.; et al. A unified nomenclature and amino acid numbering for human PTEN. Sci. Signal. 2014, 7, pe15. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Lu, J.; Jeong, H.W.; Kong, N.; Yang, Y.; Carroll, J.; Luo, H.R.; Silberstein, L.E.; Yupoma; Chai, L. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS ONE 2009, 4, e5577. [Google Scholar] [CrossRef]
- Khan, S.; Kumagai, T.; Vora, J.; Bose, N.; Sehgal, I.; Koeffler, P.H.; Bose, S. PTEN promoter is methylated in a proportion of invasive breast cancers. Int. J. Cancer 2004, 112, 407–410. [Google Scholar] [CrossRef]
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006, 66, 6546–6552. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Lee, H.Y.; Lee, J.I.; Wang, L.; Issa, J.P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 2002, 8, 1178–1184. [Google Scholar] [PubMed]
- Marsit, C.J.; Zheng, S.; Aldape, K.; Hinds, P.W.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. PTEN expression in non-small-cell lung cancer: Evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum. Pathol. 2005, 36, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Qiu, X.; Cheng, D.; Zhu, C.; Chen, L. MicroRNA-524 promotes cell proliferation by down-regulating PTEN expression in osteosarcoma. Cancer Cell Int. 2018, 18, 114. [Google Scholar] [CrossRef]
- Ramirez-Moya, J.; Wert-Lamas, L.; Santisteban, P. MicroRNA-146b promotes PI3K/AKT pathway hyperactivation and thyroid cancer progression by targeting PTEN. Oncogene 2018, 37, 3369–3383. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Huang, Y.; Gong, W. miR-205 promotes the growth, metastasis and chemoresistance of NSCLC cells by targeting PTEN. Oncol. Rep. 2013, 30, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Fang, L.; Huang, Y.; Li, R.; Yuan, J.; Yang, Y.; Zhu, X.; Chen, B.; Wu, J.; Li, M. miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer. Cancer Res. 2013, 73, 5402–5415. [Google Scholar] [CrossRef]
- Dong, L.; Li, G.; Li, Y.; Zhu, Z. Upregulation of Long Noncoding RNA GAS5 Inhibits Lung Cancer Cell Proliferation and Metastasis via miR-205/PTEN Axis. Med. Sci. Monit. 2019, 25, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.L.; Wang, H.; Liu, J.; Wang, Z.X. MicroRNA-21 (miR-21) expression promotes growth, metastasis, and chemo- or radioresistance in non-small cell lung cancer cells by targeting PTEN. Mol. Cell. Biochem. 2013, 372, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.H.; Kim, D.; Bae, S.Y.; Kim, W.K.; Hong, J.Y.; Lee, H.J.; Rajasekaran, N.; Kwon, S.; Fan, Y.; Luu, T.T.; et al. Targeting Nicotinamide N-Methyltransferase and miR-449a in EGFR-TKI-Resistant Non-Small-Cell Lung Cancer Cells. Mol. Ther. Nucleic Acids 2018, 11, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, S.; Ma, F.; Zhang, W. miRNA328 overexpression confers cisplatin resistance in nonsmall cell lung cancer via targeting of PTEN. Mol. Med. Rep. 2018, 18, 4563–4570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, B.; Sun, L.; Yan, Q.; Zhang, Y.; Zhang, Z.; Su, Y.; Wang, C. MicroRNA-130b targets PTEN to induce resistance to cisplatin in lung cancer cells by activating Wnt/beta-catenin pathway. Cell. Biochem. Funct. 2018, 36, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Perumal, E.; So Youn, K.; Sun, S.; Seung-Hyun, J.; Suji, M.; Jieying, L.; Yeun-Jun, C. PTEN inactivation induces epithelial-mesenchymal transition and metastasis by intranuclear translocation of beta-catenin and snail/slug in non-small cell lung carcinoma cells. Lung Cancer 2019, 130, 25–34. [Google Scholar] [CrossRef]
- Yu, E.J.; Hooker, E.; Johnson, D.T.; Kwak, M.K.; Zou, K.; Luong, R.; He, Y.; Sun, Z. LZTS2 and PTEN collaboratively regulate ss-catenin in prostatic tumorigenesis. PLoS ONE 2017, 12, e0174357. [Google Scholar] [CrossRef]
- Yang, W.; Bai, J.; Liu, D.; Wang, S.; Zhao, N.; Che, R.; Zhang, H. MiR-93-5p up-regulation is involved in non-small cell lung cancer cells proliferation and migration and poor prognosis. Gene 2018, 647, 13–20. [Google Scholar] [CrossRef]
- Wang, H.; Guan, X.; Tu, Y.; Zheng, S.; Long, J.; Li, S.; Qi, C.; Xie, X.; Zhang, H.; Zhang, Y. MicroRNA-29b attenuates non-small cell lung cancer metastasis by targeting matrix metalloproteinase 2 and PTEN. J. Exp. Clin. Cancer Res. 2015, 34, 59. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Gong, F.; Zhang, Y.; Jiang, J.; Zhang, H. MicroRNA-92a promotes growth, metastasis, and chemoresistance in non-small cell lung cancer cells by targeting PTEN. Tumour Biol. 2016, 37, 3215–3225. [Google Scholar] [CrossRef]
- Wang, H.; Ma, Z.; Liu, X.; Zhang, C.; Hu, Y.; Ding, L.; Qi, P.; Wang, J.; Lu, S.; Li, Y. MiR-183-5p is required for non-small cell lung cancer progression by repressing PTEN. Biomed. Pharmacother. 2019, 111, 1103–1111. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Z.; Xu, F.C.; Li, J.; Zeng, D.; Cao, X.Q.; Han, Y. MicroRNA-374b accelerates the development of lung cancer through downregulating PTEN expression via activating PI3K/Akt pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, H.; Liao, Y.; Chen, N.; Liu, T.; Zhang, H.; Zhang, H. Expression and clinical evidence of miR-494 and PTEN in non-small cell lung cancer. Tumour Biol. 2015, 36, 6965–6972. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Wang, X.; Zhu, J.; Tang, H.; Du, W.; Zeng, Y.; Sun, L.; Huang, J.A.; Liu, Z. MicroRNA-4286 promotes cell proliferation, migration, and invasion via PTEN regulation of the PI3K/Akt pathway in non-small cell lung cancer. Cancer Med. 2019. [Google Scholar] [CrossRef]
- Haddadi, N.; Lin, Y.; Travis, G.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. PTEN/PTENP1: ’Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol. Cancer 2018, 17, 37. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Kats, L.; Salmena, L.; Weiss, D.; Tan, S.M.; Ala, U.; Karreth, F.; Poliseno, L.; Provero, P.; Di Cunto, F.; et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell 2011, 147, 344–357. [Google Scholar] [CrossRef]
- Karreth, F.A.; Tay, Y.; Perna, D.; Ala, U.; Tan, S.M.; Rust, A.G.; DeNicola, G.; Webster, K.A.; Weiss, D.; Perez-Mancera, P.A.; et al. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell 2011, 147, 382–395. [Google Scholar] [CrossRef]
- Malaney, P.; Palumbo, E.; Semidey-Hurtado, J.; Hardee, J.; Stanford, K.; Kathiriya, J.J.; Patel, D.; Tian, Z.; Allen-Gipson, D.; Dave, V. PTEN Physically Interacts with and Regulates E2F1-mediated Transcription in Lung Cancer. Cell Cycle 2018, 17, 947–962. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Li, R.; Xu, X.; Zhang, L.; Lian, R.; Fang, L.; Huang, Y.; Feng, X.; Liu, X.; Li, X.; et al. CK1alpha suppresses lung tumour growth by stabilizing PTEN and inducing autophagy. Nat. Cell Biol. 2018, 20, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Nowak, D.G.; Narula, N.; Robinson, B.; Watrud, K.; Ambrico, A.; Herzka, T.M.; Zeeman, M.E.; Minderer, M.; Zheng, W.; et al. The nuclear transport receptor Importin-11 is a tumor suppressor that maintains PTEN protein. J. Cell Biol. 2017, 216, 641–656. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, S.S.; Saito, K.; Williams, S.; Arimura, Y.; Ma, Y.; Ke, Y.; Baron, V.; Mercola, D.; Feng, G.S.; et al. PTEN regulation by Akt-EGR1-ARF-PTEN axis. EMBO J. 2009, 28, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Escriva, M.; Peiro, S.; Herranz, N.; Villagrasa, P.; Dave, N.; Montserrat-Sentis, B.; Murray, S.A.; Franci, C.; Gridley, T.; Virtanen, I.; et al. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol. Cell Biol. 2008, 28, 1528–1540. [Google Scholar] [CrossRef] [PubMed]
- Uygur, B.; Abramo, K.; Leikina, E.; Vary, C.; Liaw, L.; Wu, W.S. SLUG is a direct transcriptional repressor of PTEN tumor suppressor. Prostate 2015, 75, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.B.; Li, J.; Liao, W.T.; Feng, Y.; Yu, C.P.; Hu, L.J.; Kong, Q.L.; Xu, L.H.; Zhang, X.; Liu, W.L.; et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Investig. 2009, 119, 3626–3636. [Google Scholar] [CrossRef] [Green Version]
- Hettinger, K.; Vikhanskaya, F.; Poh, M.K.; Lee, M.K.; de Belle, I.; Zhang, J.T.; Reddy, S.A.; Sabapathy, K. c-Jun promotes cellular survival by suppression of PTEN. Cell Death Differ. 2007, 14, 218–229. [Google Scholar] [CrossRef]
- Xia, D.; Srinivas, H.; Ahn, Y.H.; Sethi, G.; Sheng, X.; Yung, W.K.; Xia, Q.; Chiao, P.J.; Kim, H.; Brown, P.H.; et al. Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NF kappa B-dependent pathway. J. Biol. Chem. 2007, 282, 3507–3519. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qiu, R.; Qiu, X.; Tian, T. EYA2 promotes lung cancer cell proliferation by downregulating the expression of PTEN. Oncotarget 2017, 8, 110837–110848. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364. [Google Scholar] [CrossRef]
- Qin, J.; Wu, S.P.; Creighton, C.J.; Dai, F.; Xie, X.; Cheng, C.M.; Frolov, A.; Ayala, G.; Lin, X.; Feng, X.H.; et al. COUP-TFII inhibits TGF-beta-induced growth barrier to promote prostate tumorigenesis. Nature 2013, 493, 236–240. [Google Scholar] [CrossRef]
- Perez-Ramirez, C.; Canadas-Garre, M.; Molina, M.A.; Faus-Dader, M.J.; Calleja-Hernandez, M.A. PTEN and PI3K/AKT in non-small-cell lung cancer. Pharmacogenomics 2015, 16, 1843–1862. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Creighton, C.J.; Biswal, N.C.; Kumar, V.; Shea, M.; Herrera, S.; Contreras, A.; Gutierrez, C.; Wang, T.; Nanda, S.; et al. Overcoming endocrine resistance due to reduced PTEN levels in estrogen receptor-positive breast cancer by co-targeting mammalian target of rapamycin, protein kinase B, or mitogen-activated protein kinase kinase. Breast Cancer Res. 2014, 16, 430. [Google Scholar] [CrossRef] [PubMed]
- Lebok, P.; Kopperschmidt, V.; Kluth, M.; Hube-Magg, C.; Ozden, C.; Taskin, B.; Hussein, K.; Mittenzwei, A.; Lebeau, A.; Witzel, I.; et al. Partial PTEN deletion is linked to poor prognosis in breast cancer. BMC Cancer 2015, 15, 963. [Google Scholar] [CrossRef]
- Marsh, D.J.; Coulon, V.; Lunetta, K.L.; Rocca-Serra, P.; Dahia, P.L.; Zheng, Z.; Liaw, D.; Caron, S.; Duboue, B.; Lin, A.Y.; et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum. Mol. Genet. 1998, 7, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Shaykhiev, R.; Otaki, F.; Bonsu, P.; Dang, D.T.; Teater, M.; Strulovici-Barel, Y.; Salit, J.; Harvey, B.G.; Crystal, R.G. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol. Life Sci. 2011, 68, 877–892. [Google Scholar] [CrossRef] [PubMed]
- West, K.A.; Brognard, J.; Clark, A.S.; Linnoila, I.R.; Yang, X.; Swain, S.M.; Harris, C.; Belinsky, S.; Dennis, P.A. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J. Clin. Investig. 2003, 111, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Pathak, R.R.; Grover, A.; Malaney, P.; Quarni, W.; Pandit, A.; Allen-Gipson, D.; Dave, V. Loss of phosphatase and tensin homolog (PTEN) induces leptin-mediated leptin gene expression: Feed-forward loop operating in the lung. J. Biol. Chem. 2013, 288, 29821–29835. [Google Scholar] [CrossRef]
- Iwanaga, K.; Yang, Y.; Raso, M.G.; Ma, L.; Hanna, A.E.; Thilaganathan, N.; Moghaddam, S.; Evans, C.M.; Li, H.; Cai, W.W.; et al. Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. Cancer Res. 2008, 68, 1119–1127. [Google Scholar] [CrossRef]
- Jin, H.; Xu, C.X.; Kim, H.W.; Chung, Y.S.; Shin, J.Y.; Chang, S.H.; Park, S.J.; Lee, E.S.; Hwang, S.K.; Kwon, J.T.; et al. Urocanic acid-modified chitosan-mediated PTEN delivery via aerosol suppressed lung tumorigenesis in K-ras(LA1) mice. Cancer Gene Ther. 2008, 15, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, D.; Hashimoto, N.; Sakamoto, K.; Kohnoh, T.; Kusunose, M.; Kimura, M.; Ogata, R.; Imaizumi, K.; Kawabe, T.; Hasegawa, Y. Involvement of TGFbeta-induced phosphorylation of the PTEN C-terminus on TGFbeta-induced acquisition of malignant phenotypes in lung cancer cells. PLoS ONE 2013, 8, e81133. [Google Scholar] [CrossRef]
- Kohnoh, T.; Hashimoto, N.; Ando, A.; Sakamoto, K.; Miyazaki, S.; Aoyama, D.; Kusunose, M.; Kimura, M.; Omote, N.; Imaizumi, K.; et al. Hypoxia-induced modulation of PTEN activity and EMT phenotypes in lung cancers. Cancer Cell Int. 2016, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Kim, M.J.; Jeon, H.S.; Choi, J.E.; Kim, D.S.; Lee, E.B.; Cha, S.I.; Yoon, G.S.; Kim, C.H.; Jung, T.H.; et al. PTEN mutations and relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small cell lung cancers. Lung Cancer 2010, 69, 279–283. [Google Scholar] [CrossRef]
- Ku, B.M.; Heo, M.H.; Kim, J.H.; Cho, B.C.; Cho, E.K.; Min, Y.J.; Lee, K.H.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; et al. Molecular Screening of Small Biopsy Samples Using Next-Generation Sequencing in Korean Patients with Advanced Non-small Cell Lung Cancer: Korean Lung Cancer Consortium (KLCC-13-01). J. Pathol. Transl. Med. 2018, 52, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, R.J.; Zhang, X.; Patel, P.R.; Shelton, J.W.; Escott, C.E.; Sica, G.L.; Rossi, M.R.; Hill, C.E.; Steuer, C.E.; Pillai, R.N.; et al. Next-generation sequencing and clinical outcomes of patients with lung adenocarcinoma treated with stereotactic body radiotherapy. Cancer 2017, 123, 3681–3690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrad, M.; Roy, S.; Bittar, H.T.; Dacic, S. Next-Generation Sequencing Approach to Non-Small Cell Lung Carcinoma Yields More Actionable Alterations. Arch. Pathol. Lab. Med. 2018, 142, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Stjernstrom, A.; Karlsson, C.; Fernandez, O.J.; Soderkvist, P.; Karlsson, M.G.; Thunell, L.K. Alterations of INPP4B, PIK3CA and pAkt of the PI3K pathway are associated with squamous cell carcinoma of the lung. Cancer Med. 2014, 3, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.-J.; Zuo, X.-S.; Wang, Z.-T.; Li, F.-G.; Duan, Y.; Han, M. The clinical significance of loss of FHIT and PTEN expression in 289 patients with non-small-cell lung cancer. Transl. Cancer Res. 2016, 5, 294–301. [Google Scholar] [CrossRef]
- Yoo, S.B.; Xu, X.; Lee, H.J.; Jheon, S.; Lee, C.T.; Choe, G.; Chung, J.H. Loss of PTEN Expression is an Independent Poor Prognostic Factor in Non-small Cell Lung Cancer. J. Pathol. Transl. Med. 2011, 45, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Scrima, M.; De Marco, C.; Fabiani, F.; Franco, R.; Pirozzi, G.; Rocco, G.; Ravo, M.; Weisz, A.; Zoppoli, P.; Ceccarelli, M.; et al. Signaling networks associated with AKT activation in non-small cell lung cancer (NSCLC): New insights on the role of phosphatydil-inositol-3 kinase. PLoS ONE 2012, 7, e30427. [Google Scholar] [CrossRef]
- Spoerke, J.M.; O’Brien, C.; Huw, L.; Koeppen, H.; Fridlyand, J.; Brachmann, R.K.; Haverty, P.M.; Pandita, A.; Mohan, S.; Sampath, D.; et al. Phosphoinositide 3-kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin. Cancer Res. 2012, 18, 6771–6783. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, N.; Leduc, C.; Kohler, D.; Saieg, M.A.; John, T.; Sykes, J.; Yoshimoto, M.; Pintilie, M.; Squire, J.; Shepherd, F.A.; et al. Loss of phosphatase and tensin homolog protein expression is an independent poor prognostic marker in lung adenocarcinoma. J. Thorac. Oncol. 2012, 7, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Goncharuk, V.N.; del-Rosario, A.; Kren, L.; Anwar, S.; Sheehan, C.E.; Carlson, J.A.; Ross, J.S. Co-downregulation of PTEN, KAI-1, and nm23-H1 tumor/metastasis suppressor proteins in non-small cell lung cancer. Ann. Diagn. Pathol. 2004, 8, 6–16. [Google Scholar] [CrossRef]
- Tang, J.M.; He, Q.Y.; Guo, R.X.; Chang, X.J. Phosphorylated Akt overexpression and loss of PTEN expression in non-small cell lung cancer confers poor prognosis. Lung Cancer 2006, 51, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Thompson, J.; Fisher, H.; Zhang, P.; Huang, C.C.; Wang, L. Genomic alterations of plasma cell-free DNAs in small cell lung cancer and their clinical relevance. Lung Cancer 2018, 120, 113–121. [Google Scholar] [CrossRef]
- Lu, H.Y.; Qin, J.; Han, N.; Lei, L.; Xie, F.; Li, C. EGFR, KRAS, BRAF, PTEN, and PIK3CA mutation in plasma of small cell lung cancer patients. Onco Targets Ther. 2018, 11, 2217–2226. [Google Scholar] [CrossRef] [PubMed]
- Almodovar, K.; Iams, W.T.; Meador, C.B.; Zhao, Z.; York, S.; Horn, L.; Yan, Y.; Hernandez, J.; Chen, H.; Shyr, Y.; et al. Longitudinal Cell-Free DNA Analysis in Patients with Small Cell Lung Cancer Reveals Dynamic Insights into Treatment Efficacy and Disease Relapse. J. Thorac. Oncol. 2018, 13, 112–123. [Google Scholar] [CrossRef]
- VanderLaan, P.A.; Rangachari, D.; Mockus, S.M.; Spotlow, V.; Reddi, H.V.; Malcolm, J.; Huberman, M.S.; Joseph, L.J.; Kobayashi, S.S.; Costa, D.B. Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: Correlation with clinical outcomes. Lung Cancer 2017, 106, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Cumberbatch, M.; Tang, X.; Beran, G.; Eckersley, S.; Wang, X.; Ellston, R.P.; Dearden, S.; Cosulich, S.; Smith, P.D.; Behrens, C.; et al. Identification of a subset of human non-small cell lung cancer patients with high PI3Kbeta and low PTEN expression, more prevalent in squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Hu, C.P.; He, B.X.; Chen, X.; Lu, X.X.; Xie, M.X.; Li, W.; He, S.Y.; You, S.J.; Chen, Q. PTEN expression is a prognostic marker for patients with non-small cell lung cancer: A systematic review and meta-analysis of the literature. Oncotarget 2016, 7, 57832–57840. [Google Scholar] [CrossRef]
- Soria, J.C.; Shepherd, F.A.; Douillard, J.Y.; Wolf, J.; Giaccone, G.; Crino, L.; Cappuzzo, F.; Sharma, S.; Gross, S.H.; Dimitrijevic, S.; et al. Efficacy of everolimus (RAD001) in patients with advanced NSCLC previously treated with chemotherapy alone or with chemotherapy and EGFR inhibitors. Ann. Oncol. 2009, 20, 1674–1681. [Google Scholar] [CrossRef]
- Besse, B.; Leighl, N.; Bennouna, J.; Papadimitrakopoulou, V.A.; Blais, N.; Traynor, A.M.; Soria, J.C.; Gogov, S.; Miller, N.; Jehl, V.; et al. Phase II study of everolimus-erlotinib in previously treated patients with advanced non-small-cell lung cancer. Ann. Oncol. 2014, 25, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Owonikoko, T.K.; Behera, M.; Subramanian, J.; Saba, N.F.; Kono, S.A.; Gal, A.A.; Sica, G.; Harvey, R.D.; Chen, Z.; et al. Phase II study of docetaxel in combination with everolimus for second- or third-line therapy of advanced non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 369–372. [Google Scholar] [CrossRef]
- Deutsch, E.; Le Pechoux, C.; Faivre, L.; Rivera, S.; Tao, Y.; Pignon, J.P.; Angokai, M.; Bahleda, R.; Deandreis, D.; Angevin, E.; et al. Phase I trial of everolimus in combination with thoracic radiotherapy in non-small-cell lung cancer. Ann. Oncol. 2015, 26, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Reungwetwattana, T.; Molina, J.R.; Mandrekar, S.J.; Allen-Ziegler, K.; Rowland, K.M.; Reuter, N.F.; Luyun, R.F.; Dy, G.K.; Marks, R.S.; Schild, S.E.; et al. Brief report: A phase II “window-of-opportunity” frontline study of the MTOR inhibitor, temsirolimus given as a single agent in patients with advanced NSCLC, an NCCTG study. J. Thorac. Oncol. 2012, 7, 919–922. [Google Scholar] [CrossRef]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef]
- Yamamoto, C.; Basaki, Y.; Kawahara, A.; Nakashima, K.; Kage, M.; Izumi, H.; Kohno, K.; Uramoto, H.; Yasumoto, K.; Kuwano, M.; et al. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res. 2010, 70, 8715–8725. [Google Scholar] [CrossRef]
- Zhu, Z.; Yu, T.; Chai, Y. Multiple primary lung cancer displaying different EGFR and PTEN molecular profiles. Oncotarget 2016, 7, 81969–81971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- To, K.K.W.; Wu, W.K.K.; Loong, H.H.F. PPARgamma agonists sensitize PTEN-deficient resistant lung cancer cells to EGFR tyrosine kinase inhibitors by inducing autophagy. Eur. J. Pharmacol. 2018, 823, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Hur, G.Y.; Jung, K.H.; Jung, H.C.; Lee, S.Y.; Kim, J.H.; Shin, C.; Shim, J.J.; In, K.H.; Kang, K.H.; et al. PPAR-gamma agonist increase gefitinib's antitumor activity through PTEN expression. Lung Cancer 2006, 51, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Trotman, L.C.; Koppie, T.; Alimonti, A.; Chen, Z.; Gao, Z.; Wang, J.; Erdjument-Bromage, H.; Tempst, P.; Cordon-Cardo, C.; et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 2007, 128, 129–139. [Google Scholar] [CrossRef]
- Amodio, N.; Scrima, M.; Palaia, L.; Salman, A.N.; Quintiero, A.; Franco, R.; Botti, G.; Pirozzi, P.; Rocco, G.; De Rosa, N.; et al. Oncogenic role of the E3 ubiquitin ligase NEDD4-1, a PTEN negative regulator, in non-small-cell lung carcinomas. Am. J. Pathol. 2010, 177, 2622–2634. [Google Scholar] [CrossRef]
- Sun, H.; Ma, H.; Wang, J.; Xia, L.; Zhu, G.; Wang, Z.; Sun, J.; Chen, Z. Phosphatase and tensin homolog deleted on chromosome 10 degradation induced by NEDD4 promotes acquired erlotinib resistance in non-small-cell lung cancer. Tumour Biol. 2017, 39, 1010428317709639. [Google Scholar] [CrossRef]
- Maeda, M.; Murakami, Y.; Watari, K.; Kuwano, M.; Izumi, H.; Ono, M. CpG hypermethylation contributes to decreased expression of PTEN during acquired resistance to gefitinib in human lung cancer cell lines. Lung Cancer 2015, 87, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Diao, X.Y.; Zhang, X.; Shao, Q.; Feng, Y.F.; An, X.; Wang, H.Y. Identification of genetic alterations associated with primary resistance to EGFR-TKIs in advanced non-small-cell lung cancer patients with EGFR sensitive mutations. Cancer Commun. 2019, 39, 7. [Google Scholar] [CrossRef]
- Endoh, H.; Yatabe, Y.; Kosaka, T.; Kuwano, H.; Mitsudomi, T. PTEN and PIK3CA expression is associated with prolonged survival after gefitinib treatment in EGFR-mutated lung cancer patients. J. Thorac. Oncol. 2006, 1, 629–634. [Google Scholar]
- Milella, M.; Falcone, I.; Conciatori, F.; Matteoni, S.; Sacconi, A.; De Luca, T.; Bazzichetto, C.; Corbo, V.; Simbolo, M.; Sperduti, I.; et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci. Rep. 2017, 7, 43013. [Google Scholar] [CrossRef] [Green Version]
- Grizzi, G.; Caccese, M.; Gkountakos, A.; Carbognin, L.; Tortora, G.; Bria, E.; Pilotto, S. Putative predictors of efficacy for immune checkpoint inhibitors in non-small-cell lung cancer: Facing the complexity of the immune system. Expert Rev. Mol. Diagn. 2017, 17, 1055–1069. [Google Scholar] [CrossRef]
- Huynh, A.; DuPage, M.; Priyadharshini, B.; Sage, P.T.; Quiros, J.; Borges, C.M.; Townamchai, N.; Gerriets, V.A.; Rathmell, J.C.; Sharpe, A.H.; et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat. Immunol. 2015, 16, 188–196. [Google Scholar] [CrossRef]
- Shrestha, S.; Yang, K.; Guy, C.; Vogel, P.; Neale, G.; Chi, H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat. Immunol. 2015, 16, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.D.; Shinde, R.; McGaha, T.L.; Huang, L.; Holmgaard, R.B.; Wolchok, J.D.; Mautino, M.R.; Celis, E.; Sharpe, A.H.; Francisco, L.M.; et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci. Adv. 2015, 1, e1500845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.C.; Cao, X.; Sun, C.; Xie, Z.; Guo, J.J.; Yang, J.J.; Yang, X.N.; Dai, H.J.; Li, S.C.; Xu, X.R.; et al. Characterization of PD-L1 expression in Chinese non-small cell lung cancer patients with PTEN expression as a means for tissue quality screening. Cancer Immunol. Immunother. 2018, 67, 471–481. [Google Scholar] [CrossRef]
- Chen, C.L.; Chiang, T.H.; Tseng, P.C.; Wang, Y.C.; Lin, C.F. Loss of PTEN causes SHP2 activation, making lung cancer cells unresponsive to IFN-gamma. Biochem. Biophys. Res. Commun. 2015, 466, 578–584. [Google Scholar] [CrossRef]
- Sloot, Y.J.E.; Rabold, K.; Netea, M.G.; Smit, J.W.A.; Hoogerbrugge, N.; Netea-Maier, R.T. Effect of PTEN inactivating germline mutations on innate immune cell function and thyroid cancer-induced macrophages in patients with PTEN hamartoma tumor syndrome. Oncogene 2019, 38, 3743–3755. [Google Scholar] [CrossRef]
- Cheng, F.; Eng, C. PTEN Mutations Trigger Resistance to Immunotherapy. Trends Mol. Med. 2019. [Google Scholar] [CrossRef]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Ali, S.M.; Schrock, A.B.; Albacker, L.A.; Miller, V.A.; Stephens, P.J.; Crilley, P.; Markman, M. Response to rapamycin analogs but not PD-1 inhibitors in PTEN-mutated metastatic non-small-cell lung cancer with high tumor mutational burden. Lung Cancer 2018, 9, 45–47. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| microRNA | Expression | Material | Clinical Correlation | Ref. |
|---|---|---|---|---|
| miR-21 | High | Tissue/in vitro | Chemo- and radio resistance | [29] |
| miR-29b | Low | Tissue/in vitro | LN metastasis | [36] |
| miR-92a | High | Tissue | Tumor stage/LN metastasis | [37] |
| miR-93-5p | High | Tissue | Poor survival | [35] |
| miR-130b | High | In vitro/In vivo | Cisplatin resistance | [32] |
| miR-183-5p | High | Tissue/in vitro | Tumor volume | [38] |
| miR-205 | High | Tissue/in vitro | Chemo resistance | [26,27,28] |
| miR-328 | High | Tissue/in vitro | Cisplatin resistance | [31] |
| miR-374b | High | Tissue/in vitro | NR | [39] |
| miR-449a | Low | EGFR TKI res NSCLC tissue/in vitro/in vivo | EGFR TKI resistance | [30] |
| miR-494 | High | Tissue | LN metastasis/poor OS | [40] |
| miR-4286 | High | Tissue/in vitro | Histology | [41] |
| Histologic Type | Finding | Number of Patients | Human Material | Correlated Parameters | Discovery Technique | Ref. |
|---|---|---|---|---|---|---|
| NSCLC | Protein loss, Promoter methylation | 24% (30/125), 35% (7/20) | Tissue | NR | IHC, PCR | [22] |
| NSCLC | Promoter methylation | 26% (39/151) | Tissue | No predictor of protein expression | PCR | [23] |
| NSCLC | Mutation | 4.5% (8/176) | Tissue | smokers, mostly SQLC | PCR, sequencing assays | [69] |
| A-NSCLC | Mutation | 2.5% (4/162) | Tissue | NR | NGS | [70] |
| LUAD | Mutation | 2.2% (1/45) | Tissue | NR | NGS | [71] |
| LUAD | Deletion | 6.8% (2/29) | Tissue | NR | NGS | [72] |
| NSCLC | Protein loss, mutation | 50% (86/173), 4% (7/180) | Tissue | NR | IHC, PCR | [73] |
| NSCLC | Protein loss, weak | 44% (52/117), 29% (34/117) | Tissue | Stage I and II | IHC | [22] |
| NSCLC | Protein loss | 59.86% (173/289) | Tissue | LN metastasis Smoking status, Decreased survival | IHC | [74] |
| NSCLC | Protein loss | 42.4% (122/288) | Tissue | SQLC, Smoking status; Decreased PFS | IHC | [75] |
| NSCLC | Protein loss | 39% (41/104) | Tissue | More prevalent in SQLC | IHC | [76] |
| SQLC, LUAD | Protein loss | 21% (9/43), 4% (2/56) | Tissue | PTEN gene loss was associated with SQLC | IHC | [77] |
| NSCLC | Protein loss, Deletion | 41.4% (63/152), 5.6% (7/124) | Tissue | More prevalent in SQLC, shorter DFS for LUAD | IHC, FISH | [78] |
| NSCLC | Protein loss | 41.4% (43/104) | Tissue | Advanced disease, LN metastasis, Decreased survival | IHC | [79] |
| NSCLC | Protein loss | 46.1% (47/102) | Tissue | Poor survival for p-AktS473 positive and PTEN negative | IHC | [80] |
| SCLC | Deletion | 29% (7/24) | cf-DNA | NR | WGS | [81] |
| SCLC | Mutation | 0% (0/99) | cf-DNA | NR | HRM | [82] |
| SCLC | Mutation | 7.4% (2/27) | cf-DNA | NR | NGS | [83] |
| Agent | Target | Phase | Setting | Main Results | Ref. |
|---|---|---|---|---|---|
| Everolimus | mTORC1 | II | Pretreated NSCLC | DCR 47.1%; mPFS 2.6–2.7 months | [87] |
| Everolimus (+ erl vs. erl) | mTORC1 | IIR | Pretreated NSCLC | mPFS 2.9 vs. 2.0 months; G3/4 AEs 72.7% vs. 32.3% | [88] |
| Everolimus (+ docetaxel) | mTORC1 | II | Pretreated NSCLC | 6-month PFS 5%, mOS 9.6 months | [89] |
| Everolimus (+ thoracic RT) | mTORC1 | I | Locally advanced or metastatic untreated NSCLC | Reccommended dose with RT 50mg/week; relevant pulmonary AEs | [90] |
| Temsirolimus | mTORC1 | II | Advanced NSCLC | mPFS 23 months, mOS 6.6 months | [91] |
| Vistusertib | mTORC1/2 | II | Advanced RICTOR-amplified SCLC | Ongoing | NCT03106155 |
| MK-2206 | Pan-AKT | II | PI3KCA, AKT and PTEN-mutant NSCLC and SCLC | Ongoing | NCT01306045 |
| Gedatolisib (+ carbo-paclitaxel) | Dual PI3K and mTORC1/2 | I/II | Pretreated NSCLC | Ongoing | NCT02920450 |
| Gedatolisib (+ palbociclib) | Dual PI3K and mTORC1/2 | I | Squamous pretreated NSCLC | Ongoing | NCT03065062 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gkountakos, A.; Sartori, G.; Falcone, I.; Piro, G.; Ciuffreda, L.; Carbone, C.; Tortora, G.; Scarpa, A.; Bria, E.; Milella, M.; et al. PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around. Cancers 2019, 11, 1141. https://doi.org/10.3390/cancers11081141
Gkountakos A, Sartori G, Falcone I, Piro G, Ciuffreda L, Carbone C, Tortora G, Scarpa A, Bria E, Milella M, et al. PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around. Cancers. 2019; 11(8):1141. https://doi.org/10.3390/cancers11081141
Chicago/Turabian StyleGkountakos, Anastasios, Giulia Sartori, Italia Falcone, Geny Piro, Ludovica Ciuffreda, Carmine Carbone, Giampaolo Tortora, Aldo Scarpa, Emilio Bria, Michele Milella, and et al. 2019. "PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around" Cancers 11, no. 8: 1141. https://doi.org/10.3390/cancers11081141
APA StyleGkountakos, A., Sartori, G., Falcone, I., Piro, G., Ciuffreda, L., Carbone, C., Tortora, G., Scarpa, A., Bria, E., Milella, M., Rosell, R., Corbo, V., & Pilotto, S. (2019). PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around. Cancers, 11(8), 1141. https://doi.org/10.3390/cancers11081141