The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2− Metastatic Breast Cancer: Biological Mechanisms and New Treatments


Abstract
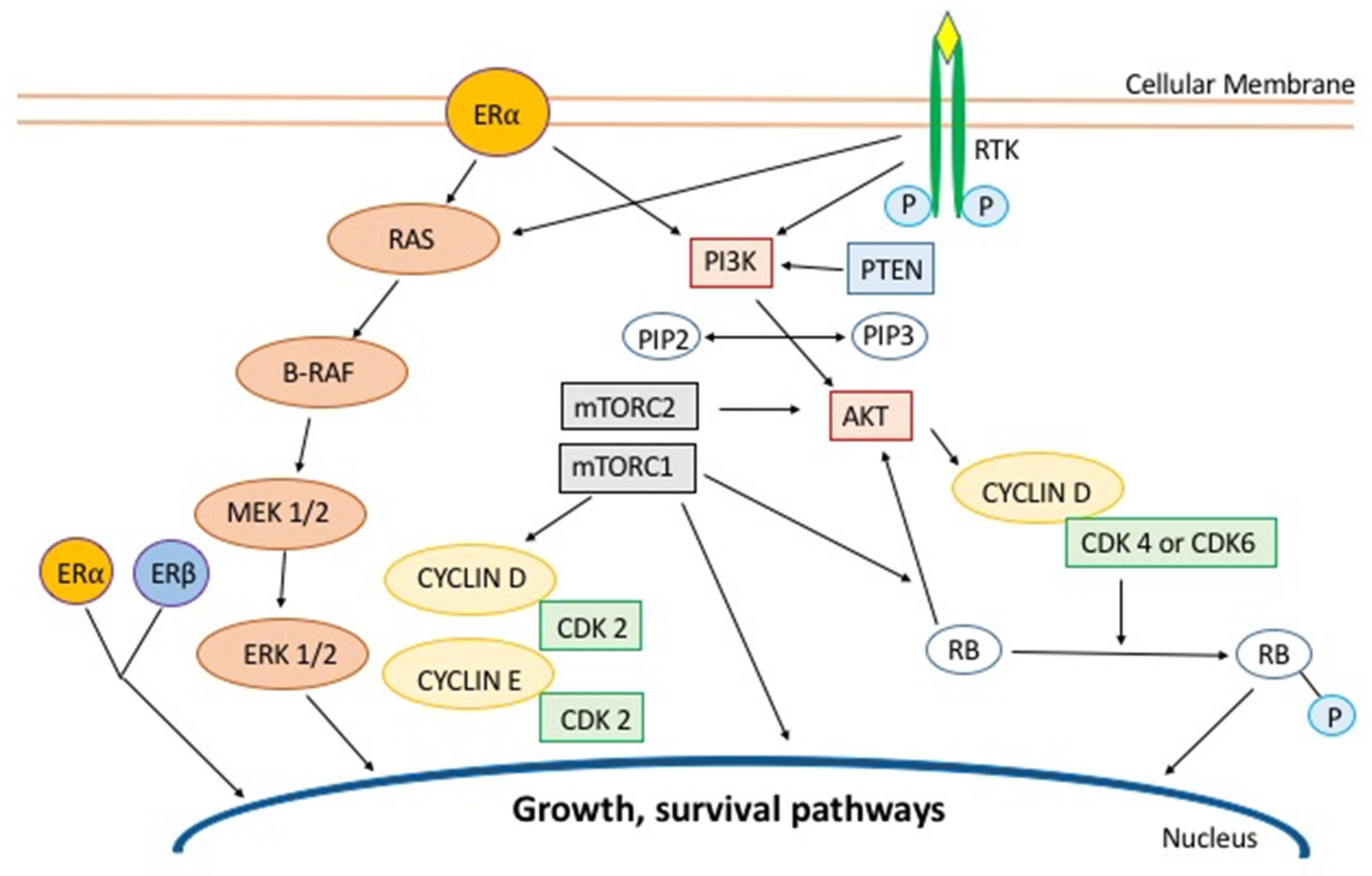
:1. Introduction
2. Biological Mechanisms behind Endocrine Resistance
3. PI3K/AKT/mTOR Pathway
3.1. mTOR
3.1.1. From Biology to Drug Development
3.1.2. Clinical Trials
3.1.3. Mechanisms of Endocrine Resistance during mTOR Inhibition
3.2. PI3K/AKT
3.2.1. The Intracellular Molecular Pathway
3.2.2. Clinical Trials
3.2.3. Mechanisms of Endocrine Resistance in PI3K/AKT Pathway
4. CDK4/6 Inhibitors
4.1. Biological Functions and Cross-Link Interactions
4.2. Clinical Trials
4.2.1. Palbociclib
4.2.2. Ribociclib
4.2.3. Abemaciclib
4.3. Mechanisms of Endocrine Resistance in the CDK 4/6 Pathway
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; André, F.; Harbeck, N.; Aguilar Lopez, B.; Barrios, C.H.; Bergh, J.; et al. 4th ESO–ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4)†. Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Quan, J.; Yang, J.; Chen, Z. The potential markers of endocrine resistance among HR+ /HER2+ breast cancer patients. Clin. Transl. Oncol. 2019. [Google Scholar] [CrossRef]
- O’Leary, B.; Cutts, R.; Huang, X.; Hrebien, S.; Liu, Y.; Garcia-Murillas, I.; Andre, F.; Loi, S.; Loibl, S.; Cristofanilli, M.; et al. Genomic markers of early progression on fulvestrant with or without palbociclib for ER+ advanced breast cancer in the PALOMA-3 trial. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef]
- Eyster, K.M. The Estrogen Receptors: An Overview from Different Perspectives. Methods Mol. Biol. 2016, 1366, 1–10. [Google Scholar] [CrossRef]
- Arnal, J.F.; Fontaine, C.; Abot, A.; Valera, M.C.; Laurell, H.; Gourdy, P.; Lenfant, F. Lessons from the dissection of the activation functions (AF-1 and AF-2) of the estrogen receptor alpha in vivo. Steroids 2013, 78, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Bostner, J.; Skoog, L.; Fornander, T.; Nordenskjold, B.; Stal, O. Estrogen receptor-alpha phosphorylation at serine 305, nuclear p21-activated kinase 1 expression, and response to tamoxifen in postmenopausal breast cancer. Clin. Cancer Res. 2010, 16, 1624–1633. [Google Scholar] [CrossRef]
- Rani, A.; Stebbing, J.; Giamas, G.; Murphy, J. Endocrine Resistance in Hormone Receptor Positive Breast Cancer-From Mechanism to Therapy. Front. Endocrinol. 2019, 10, 245. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, T.J.; Meier, J.B.; Jansen, V.M. Current Landscape of Targeted Therapies for Hormone-Receptor Positive, HER2 Negative Metastatic Breast Cancer. Front. Oncol. 2018, 8, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef]
- Cortés, J.; Im, S.A.; Holgado, E.; Perez-Garcia, J.M.; Schmid, P.; Chavez-MacGregor, M. The next era of treatment for hormone receptor-positive, HER2-negative advanced breast cancer: Triplet combination-based endocrine therapies. Cancer Treat. Rev. 2017, 61, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Lange, C.A.; Yee, D. Killing the second messenger: Targeting loss of cell cycle control in endocrine-resistant breast cancer. Endocr. Relat. Cancer 2011, 18, 19–24. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Jacquier, M.; Kuriakose, S.; Bhardwaj, A.; Zhang, Y.; Shrivastav, A.; Portet, S.; Varma Shrivastav, S. Investigation of Novel Regulation of N-myristoyltransferase by Mammalian Target of Rapamycin in Breast Cancer Cells. Sci. Rep. 2018, 8, 12969. [Google Scholar] [CrossRef]
- Thangavel, C.; Dean, J.L.; Ertel, A.; Knudsen, K.E.; Aldaz, C.M.; Witkiewicz, A.K.; Clarke, R.; Knudsen, E.S. Therapeutically activating RB: Reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr. Relat. Cancer 2011, 18, 333–345. [Google Scholar] [CrossRef]
- Pronzato, P. Role of everolimus in the treatment of metastatic HER2-negative/HR-positive breast cancer. Future Oncol. 2017, 13, 1371–1384. [Google Scholar] [CrossRef]
- Massacesi, C.; Di Tomaso, E.; Urban, P.; Germa, C.; Quadt, C.; Trandafir, L.; Aimone, P.; Fretault, N.; Dharan, B.; Tavorath, R.; et al. PI3K inhibitors as new cancer therapeutics: Implications for clinical trial design. Onco Targets Ther. 2016, 9, 203–210. [Google Scholar] [CrossRef]
- Spring, L.; Bardia, A.; Modi, S. Targeting the cyclin D-cyclin-dependent kinase (CDK) 4/6-retinoblastoma pathway with selective CDK 4/6 inhibitors in hormone receptor-positive breast cancer: Rationale, current status, and future directions. Discov. Med. 2016, 21, 65–74. [Google Scholar]
- Baker, S.J.; Reddy, E.P. CDK4. A Key Player in the Cell Cycle, Development, and Cancer. Genes Cancer 2012, 3, 658–669. [Google Scholar] [CrossRef]
- Dey, N.; De, P.; Leyland-Jones, B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017, 175, 91–106. [Google Scholar] [CrossRef]
- Yardley, D.A.; Noguchi, S.; Pritchard, K.I.; Burris, H.A., 3rd; Baselga, J.; Gnant, M.; Hortobagyi, G.N.; Campone, M.; Pistilli, B.; Piccart, M.; et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv. Ther. 2013, 30, 870–884. [Google Scholar] [CrossRef]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.C.; Debled, M.; Spaëth, D.; et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: A GINECO study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef]
- Piccart, M.; Hortobagyi, G.N.; Campone, M.; Pritchard, K.I.; Lebrun, F.; Ito, Y.; Noguchi, S.; Perez, A.; Rugo, H.S.; Deleu, I.; et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Overall survival results from BOLERO-2. Ann. Oncol. 2014, 25, 2357–2362. [Google Scholar] [CrossRef]
- Royce, M.; Bachelot, T.; Villanueva, C.; Özgüroglu, M.; Azevedo, S.J.; Cruz, F.M.; Debled, M.; Hegg, R.; Toyama, T.; Falkson, C.; et al. Everolimus Plus Endocrine Therapy for Postmenopausal Women with Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: A Clinical Trial. JAMA Oncol. 2018, 4, 977–984. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef]
- Cazzaniga, M.E.; Airoldi, M.; Arcangeli, V.; Artale, S.; Atzori, F.; Ballerio, A.; Bianchi, G.V.; Blasi, L.; Campidoglio, S.; Ciccarese, M.; et al. On behalf of EVA Study Group. Efficacy and safety of Everolimus and Exemestane in hormone-receptor positive (HR+) human-epidermal-growth-factor negative (HER2-) advanced breast cancer patients: New insights beyond clinical trials. The EVA study. Breast 2017, 35, 115–121. [Google Scholar] [CrossRef]
- Schmid, P.; Zaiss, M.; Harper-Wynne, C.; Ferreira, M.; Dubey, S.; Chan, S.; Makris, A.; Nemsadze, G.; Brunt, A.M.; Kuemmel, S.; et al. Abstract GS2-07 MANTA-A randomized phase II study of fulvestrant in combination with the dual mTOR inhibitor AZD2014 or everolimus or fulvestrant alone in estrogen receptor-positive advanced or metastatic breast cancer. Cancer Res. 2018, 78. [Google Scholar] [CrossRef]
- Faes, S.; Planche, A.; Uldry, E.; Santoro, T.; Pythoud, C.; Stehle, J.C.; Horlbeck, J.; Letovanec, I.; Riggi, N.; Dattaet, D.; et al. Targeting carbonic anhydrase IX improves the anti-cancer efficacy of mTOR inhibitors. Oncotarget 2016, 7, 36666–36680. [Google Scholar] [CrossRef]
- Faes, S.; Demartines, N.; Dormond, O. Resistance to mTORC1 Inhibitors in Cancer Therapy: From Kinase Mutations to Intratumoral Heterogeneity of Kinase Activity. Oxid. Med. Cell. Longev. 2017, 2017, 1726078. [Google Scholar] [CrossRef]
- De Iuliis, F.; Salerno, G.; Giuffrida, A.; Milana, B.; Taglieri, L.; Rubinacci, G.; Giantulli, S.; Terella, F.; Silvestri, I.; Scarpa, S. Breast cancer cells respond differently to docetaxel depending on their phenotype and on survivin upregulation. Tumor Biol. 2016, 37, 2603–2611. [Google Scholar] [CrossRef]
- Taglieri, L.; De Iuliis, F.; Giuffrida, A.; Giantulli, S.; Silvestri, I.; Scarpa, S. Resistance to the mTOR inhibitor everolimus is reversed by the downregulation of survivin in breast cancer cells. Oncol. Lett. 2017, 14, 3832–3838. [Google Scholar] [CrossRef] [Green Version]
- Bihani, T.; Ezell, S.A.; Ladd, B.; Grosskurth, S.E.; Mazzola, A.M.; Pietras, M.; Reimer, C.; Zinda, M.; Fawell, S.; D’Cruz, C.M. Resistance to everolimus driven by epigenetic regulation of MYC in ER+ breast cancers. Oncotarget 2014, 6, 2407–2420. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M.; Hanamura, T.; Tsuboi, K.; Kaneko, Y.; Yamaguchi, Y.; Niwa, T.; Narui, K.; Endo, I.; Hayashi, S.I. Acquired resistance to everolimus in aromatase inhibitor-resistant breast cancer. Oncotarget 2018, 9, 21468–21477. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signaling. Nat. Rev. Mol. Cell. Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Balselga, J. Targeting the Phosphoinositide-3 (PI3) Kinase Pathway in Breast Cancer. Oncologist 2011, 16, 12–19. [Google Scholar] [CrossRef]
- Mukohara, T. PI3K mutations in breast cancer: Prognostic and therapeutic implications. Breast Cancer (Dove Med Press) 2015, 7, 111–123. [Google Scholar] [CrossRef]
- Ciruelos Gil, E.V. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat. Rev. 2014, 40, 862–871. [Google Scholar] [CrossRef]
- Miller, T.W.; Hennessy, B.T.; González-Angulo, A.M.; Fox, E.M.; Mills, G.B.; Chen, H.; Higham, C.; García-Echeverría, C.; Shyr, Y.; Arteaga, C.L. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J. Clin. Investig. 2010, 120, 2406–2413. [Google Scholar] [CrossRef]
- Baselga, J.; Im, S.A.; Iwata, H.; Cortés, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Campone, M.; Im, S.A.; Iwata, H.; Clemons, M.; Ito, Y.; Awada, A.; Chia, S.; Jagiełło-Gruszfeld, A.; Pistilli, B.; Tseng, L.M.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant for postmenopausal, hormone receptor-positive, human epidermal growth factor receptor 2-negative, advanced breast cancer: Overall survival results from BELLE-2. Eur. J. Cancer 2018, 103, 147–154. [Google Scholar] [CrossRef]
- Krop, I.E.; Mayer, I.A.; Ganju, V.; Dickler, M.; Johnston, S.; Morales, S.; Yardley, D.A.; Melichar, B.; Forero-Torres, A.; Lee, S.C.; et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 811–821. [Google Scholar] [CrossRef]
- Jia, S.; Roberts, T.M.; Zhao, J.J. Should individual PI3 kinase isoforms be targeted in cancer? Curr. Opin. Cell. Biol. 2009, 21, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef]
- Juric, D.; Janku, F.; Rodón, J.; Burris, H.A.; Mayer, I.A.; Schuler, M.; Seggewiss-Bernhardt, R.; Gil-Martin, M.; Middleton, M.R.; Baselga, J.; et al. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor–Positive Advanced Breast Cancer: A Phase 1b Clinical Trial. JAMA Oncol. 2019, 5, e184475. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.M.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): Results of the phase 3 SOLAR-1 trial. In Proceedings of the ESMO 2018 Congress, Munich, Germany, 19–23 October 2018. [Google Scholar]
- Dickler, M.N.; Saura, C.; Richards, D.A.; Krop, I.E.; Cervantes, A.; Bedard, P.L.; Patel, M.R.; Pusztai, L.; Oliveira, M.; Cardenas, A.K.; et al. A Phase II Study of Taselisib (GDC-0032) in Combination with Fulvestrant in Patients with HER2-Negative, Hormone Receptor–Positive Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 4380–4387. [Google Scholar] [CrossRef]
- Baselga, J.; Dent, S.F.; Cortés, J.; Im, Y.H.; Diéras, V.; Harbeck, N.; Krop, I.E.; Verma, S.; Wilson, T.R.; Jin, H.; et al. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): Primary analysis from SANDPIPER. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Jones, R.H.; Carucci, M.; Casbard, A.C.; Butler, R.; Alchami, F.; Bale, C.J.; Bezecny, P.; Joffe, J.; Moon, S.; Twelves, C.; et al. Capivasertib (AZD5363) plus fulvestrant versus placebo plus fulvestrant after relapse or progression on an aromatase inhibitor in metastatic ER-positive breast cancer (FAKTION): A randomized, double-blind, placebo-controlled, phase II trial. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Gao, X.; Qin, T.; Mao, J.; Zhang, J.; Fan, S.; Lu, Y.; Sun, Z.; Zhang, Q.; Song, B.; Li, L. PTENP1/miR-20a/PTEN axis contributes to breast cancer progression by regulating PTEN via PI3K/AKT pathway. J. Exp. Clin. Cancer Res. 2019, 38, 256. [Google Scholar] [CrossRef]
- Chagpar, R.B.; Links, P.H.; Pastor, M.C.; Furber, L.A.; Hawrysh, A.D.; Chamberlain, M.D.; Andersonet, D.H. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA. 2010, 107, 5471–5476. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.T.; Chi, M.N.; Yang, R.H.; Guo, X.Y.; Zan, L.K.; Wang, C.Y.; Xi, Y.F.; Jin, L.; Croft, A.; Tseng, H.Y.; et al. INPP4B is an oncogenic regulator in human colon cancer. Oncogene 2016, 35, 3049–3061. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5, 308–324. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2015, 6, 353–367. [Google Scholar] [CrossRef]
- Peyressatre, M.; Prével, C.; Pellerano, M.; Morris, M.C. Targeting Cyclin-Dependent Kinases in Human Cancers: From Small Molecules to Peptide Inhibitors. Cancers 2015, 7, 179–237. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Knudsen, E.S. Retinoblastoma tumor suppressor pathway in breast cancer: Prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res. 2014, 16, 207. [Google Scholar] [CrossRef]
- Liu, Y.; Zhong, X.; Wan, S.; Zhang, W.; Lin, J.; Zhang, P.; Li, Y. p16INK4a expression in retinoblastoma: A marker of differentiation grade. Diagn. Pathol. 2014, 9, 180. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16(ink4a) expression in tumors: Functional significance, clinical associations and future developments. Cell Cycle 2011, 10, 2497–2503. [Google Scholar] [CrossRef]
- Bower, J.J.; Vance, L.D.; Psioda, M.; Smith-Roe, S.L.; Simpson, D.A.; Ibrahim, J.G.; Hoadley, K.A.; Perou, C.M.; Kaufmann, W.K. Patterns of cell cycle checkpoint deregulation associated with intrinsic molecular subtypes of human breast cancer cells. Breast Cancer 2013, 3, 9. [Google Scholar] [CrossRef]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; De Michele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Juric, D.; Munster, P.; Campone, M.; Ismail-Khan, R.; García-Estevez, L.; Hamilton, E.P.; Becerra, C.; De Boer, R.H.; Hui, R.; Goncalveset, A.; et al. Ribociclib (LEE011) and letrozole in estrogen receptor-positive (ER+), HER2-negative (HER2–) advanced breast cancer (aBC): Phase Ib safety, preliminary efficacy and molecular analysis. In Proceedings of the American Society of Clinical Oncology (ASCO) Annual Meeting, Chicago, IL, USA, 3–7 June 2016. [Google Scholar]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.L.; Winer, E.P.; et al. Updated results from MONALEESA-2, a phase 3 trial of first-line ribociclib + letrozole in hormone receptor-positive (HR+), HER2-negative (HER2–), advanced breast cancer (ABC). J. Clin. Oncol. 2017, 35, 1038. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Val Bianchi, G.; Esteva, F.J.; Martín, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef]
- Tripathy, D.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef]
- Im, S.A.; Lu, Y.S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; DeMichele, A.; Clark, A.S.; Zelnak, A.B.; Yardley, D.A.; Karuturi, M.S.; Sanft, T.B.; Blau, S.; Hart, L.L.; et al. Triplet therapy (continuous ribociclib, everolimus, exemestane) in HR+/HER2− advanced breast cancer postprogression on a CDK4/6 inhibitor (TRINITI-1): Efficacy, safety, and biomarker results. J. Clin. Oncol. 2019, 37, 1016. [Google Scholar] [CrossRef]
- Lallena, M.J.; Boehnke, K.; Torres, R.; Hermoso, A.; Amat, J.; Calsina, B.; De Dios, A.; Buchanan, S.; Du, J.; Beckmann, R.P.; et al. In-vitro characterization of Abemaciclib pharmacology in ER+ breast cancer cell lines. In Proceedings of the 106th Annual Meeting of the American Association for Cancer Research, Philadelphia, PA, USA, 18–22 April 2015; p. 3101. [Google Scholar]
- Patnaik, A.R.L.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; Nasir, A.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non–Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR+/HER2- Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 25, 2875–2884. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib as Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Johnston, S.; Martin, M.; Di Leo, A.; Im, S.A.; Awada, A.; Forrester, T.; Frenzel, M.; Hardebeck, M.C.; Cox, J.; Barriga, S.; et al. MONARCH 3 final PFS: A randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 2019, 5, 5. [Google Scholar] [CrossRef]
- Anders, C.K.; Le Rhun, E.; Bachelot, T.D.; Yardley, D.A.; Awada, A.; Conte, P.; Kabos, P.; Bear, M.; Yang, Z.; Chen, Y.; et al. A phase II study of abemaciclib in patients (pts) with brain metastases (BM) secondary to HR+, HER2- metastatic breast cancer (MBC). J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Ribnikar, D.; Volovat, S.R.; Cardoso, F. Targeting CDK4/6 pathways and beyond in breast cancer. Breast 2019, 43, 8–17. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar] [CrossRef] [Green Version]
- Teh, J.L.F.; Aplin, A.E. Arrested Developments: CDK4/6 Inhibitor Resistance and Alterations in the Tumor Immune Microenvironment. Clin Cancer Res. 2019, 25, 921–927. [Google Scholar] [CrossRef]
- Li, W.; Kotoshiba, S.; Berthet, C.; Hilton, M.B.; Kaldis, P. Rb/Cdk2/Cdk4 triple mutant mice elicit an alternative mechanism for regulation of the G1/S transition. Proc. Natl. Acad. Sci. USA 2009, 106, 486–491. [Google Scholar] [CrossRef]
- Razavi, P.; Henrique dos Anjos, C.; Brown, D.N.; Qing, L.; Ping, C.; Herbert, J.; Colon, J.; Liu, D.; Mao, M.; Norton, L.; et al. Molecular profiling of ER+ metastatic breast cancers to reveal association of genomic alterations with acquired resistance to CDK4/6 inhibitors. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef]
- Condorelli, R.; Spring, L.; O’Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 mutations and acquired resistance to CDK4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef]


{kind=link}
| Author, Year [Ref] | Trial | Study Design | N° Patients | Treatment Line | Drug | Primary Endpoints | Results |
|---|---|---|---|---|---|---|---|
| Bachelot, 2012 [23] | TAMRAD | Phase II Randomized 1:1 | 111 | Progressed on previous ET | EVE plus TAM versus TAM | CBR | 61.1% versus 42.1% |
| Yardley, 2013 [22] | BOLERO-2 | Phase III Placebo-controlled Double-blind Randomized 2:1 | 724 | Progressed on previous ET | EVE plus EXE versus PBO plus EXE | PFS | 11.0 versus 4.1 mo, p < 0.0001 |
| Baselga, 2017 [40] | BELLE-2 | Phase III Placebo-controlled Double-blind Randomized 1:1 | 1147 | Progressed on previous ET | BUP plus F500 versus PBO plus F500 | PFS (overall population and inactivated or non-activated PI3K pathway) | Overall population (n = 1147) 6.9 versus 5.0 mo, p = 0.00021; PIK3CA mutant (n = 200) 7.0 versus 3.2 mo, p = 0.0005 PIK3CA WT (n = 387) 6.8 versus 6.8 mo, p = 0.642 PI3K activated (n = 372) 6.8 versus 4.0 mo, p = 0.014 PI3K non-activated (n = 479) NR |
| Krop, 2016 [42] | FERGI | Phase II Placebo-controlled Double-blind Randomized: 1:1 (part 1) 2:1 (part 2) | 168 (part 1) 61 (part 2) | Progressed on previous ET | PIC plus F500 versus PBO plus F500 | PFS (overall population and in patients with PI3K mutated tumors) | Part 1 PIK3CA mutant (n = 70) 6.5 versus 5.1 mo, p = 0.268 PIK3CA WT (n = 84) 5.8 versus 3.6 mo, p = 0.23 Part 2 PIK3CA mutant (n = 61) 5.4 versus 10.0 mo, p = 0.84 |
| Andrè, 2018 [46] | SOLAR-1 | Phase III Double-blind Placebo-controlled Randomized 1:1 | 572 | Progressed on previous ET | ALP plus F500 versus PBO plus F500 | PFS | PIK3CA-mutated (n = 341) 11.0 versus 5.7 mo, p < 0.001 |
| Baselga, 2018 [48] | SANDPIPER | Phase III Double-blind Placebo-controlled Randomized 2:1 | 631 | Progressed on previous ET | TAS plus F500 versus PBO plus F500 | PFS | 7.4 versus 5.4 mo, p = 0.0037 |
| Author, Year [Ref] | Trial | Study Design | N° Patients | Treatment Line | Drug | Primary Endpoints | Results |
|---|---|---|---|---|---|---|---|
| Finn, 2015 [62] | PALOMA-1/ TRIO-18 | Phase II Open-label Randomized 1:1 | 165 | 1° line | LET plus PAL versus LET | PFS | 20.2 versus 10.2 mo; p = 0.0004 |
| Finn, 2016 [63] | PALOMA-2 | Phase III Placebo-controlled Double-blind Randomized 2:1 | 666 | 1° line | LET plus PAL versus LET plus PBO | PFS | 24.8 versus 14.5 mo; p < 0.001 |
| Cristofanilli, 2016 [64] | PALOMA-3 | Phase III Placebo-controlled Double-blind Randomized 2:1 | 521 | Progressed on previous ET | F500 +/-LHRH analogue plus PAL versus F500 +/-LHRH analogue plus PBO | PFS | 9.5 versus 4.6 mo; p < 0.0001 |
| Hortobagyi, 2017 [67] | MONALEESA-2 | Phase III Placebo-controlled Double-blind Randomized 1:1 | 668 | 1° line postmenopausal | LET plus RIB versus LET plus PBO | PFS | 25.3 versus 16 mo; p < 0.001 |
| Slamon, 2018 [68] | MONALEESA-3 | Phase III Placebo-controlled Double-blind Randomized 2:1 | 726 | Progressed on previous ET | F500 plus RIB versus F500 plus PBO | PFS | 20.5 versus 12.8 mo; p < 0.001 |
| Tripathy, 2018 [69] | MONALEESA-7 | Phase III Placebo-controlled Double-blind Randomized 1:1 | 672 | 1° line premenopausal | TAM/LET/ANA plus LHRH analogue plus RIB versus TAM/LET/ANA plus LHRH analogue plus PBO | PFS | 23.8 versus 13 mo; p < 0.0001 |
| Dickler, 2017 [74] | MONARCH-1 | Phase II Single agent Open-label | 132 | Progressed on previous ET | ABE | ORR | CR 0 PR 17.4 % SD 40.2 % PD 25.0% |
| Goetz, 2017 [76] | MONARCH-3 | Phase III Placebo-controlled Double-blind Randomized 2:1 | 493 | 1° line | LET or ANA plus ABE versus LET or ANA plus PBO | PFS | 28.18 versus 14.76 mo; p = 0.000002 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Presti, D.; Quaquarini, E. The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2− Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers 2019, 11, 1242. https://doi.org/10.3390/cancers11091242
Presti D, Quaquarini E. The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2− Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers. 2019; 11(9):1242. https://doi.org/10.3390/cancers11091242
Chicago/Turabian StylePresti, Daniele, and Erica Quaquarini. 2019. "The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2− Metastatic Breast Cancer: Biological Mechanisms and New Treatments" Cancers 11, no. 9: 1242. https://doi.org/10.3390/cancers11091242
APA StylePresti, D., & Quaquarini, E. (2019). The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2− Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers, 11(9), 1242. https://doi.org/10.3390/cancers11091242